
Abstract
Bile acids have important roles in the regulation of lipid, glucose and energy metabolism. Metabolic diseases linked to obesity, including type 2 diabetes mellitus and non-alcoholic fatty liver disease, are associated with dysregulation of bile acid homeostasis. Here, the basic chemistry and regulation of bile acids as well as their metabolic effects will be reviewed. Changes in circulating bile acids associated with obesity and related diseases will be reviewed. Finally, pharmaceutical manipulation of bile acid homeostasis as therapy for metabolic diseases will be outlined.
Keywords
Introduction
There is a close two-way relationship between bile acid homeostasis and nutrition. The synthesis, secretion and cycling of bile acids are governed by timing and content of food ingestion, and bile acid signalling regulates the subsequent metabolism of available fuel. Interest in bile acid biology has increased in recent years, due to global public health crises of obesity and related metabolic disorders. The number of people with obesity worldwide is more than 650 million, according to a 2016 estimate by the World Health Organization, 1 and in the UK alone, there are six million people with type 2 diabetes (T2DM). 2 Other metabolic diseases that are directly related to the pandemic of obesity include hypercholesterolaemia and non-alcoholic fatty liver disease (NAFLD). NAFLD, which is widely considered the hepatic manifestation of the metabolic syndrome, has an estimated global prevalence of 25% 3 and is strongly associated with both obesity and T2DM. 3
Increased appreciation of bile acids as metabolic regulators has led to two major research questions. Firstly, how does bile acid homeostasis change in metabolic disease? Secondly, can bile acid activity be harnessed to treat metabolic disease? Both will be addressed in this review.
Bile acid chemistry
Bile acid structure
Bile salts, which include bile acids and bile alcohols, have a central sterol ring. 4 Beyond this common feature, bile salts comprise a vast and varied family of small molecules. The bile of each species is comprised of multiple different salts, with tremendous diversity in the resulting bile salt profile among vertebrates. 5 This diversity likely represents parallel evolution of multiple biochemical pathways all serving to convert cholesterol, a crucial cellular component, but a hydrophobic and poorly soluble lipid, into a water-soluble molecule.4–6
Bile acids are planar amphipathic molecules with a carboxyl tail. 7 On one surface they display a hydroxyl group and on the other they project hydrophobic methyl groups. 8 The overall polarity and solubility depend on the bile acid species and its chemical structure. At low concentrations, bile acids dissolve in water, whereas at high concentrations, they self-associate to form aggregates, also known as micelles. This occurs because hydrophobic faces of multiple molecules line up to repel water, while the hydrophilic faces become oriented towards the surrounding water. 8
Primary bile acid biosynthesis
In mammals, there are two major biosynthetic pathways to commence formation of bile acids: ‘classic’ or ‘neutral’, and ‘alternate’ or ‘acidic’. 9 Synthesis of the majority of bile acids involves the classic pathway in which hydroxylation of the cholesterol steroid nucleus is performed by cholesterol 7α-hydroxylase (CYP7A1), the rate-limiting enzyme in the pathway.4,10 This enzyme is a member of the P450 enzyme family and is a liver-specific, microsomal mono-oxidase. Ninety per cent of mice that lack CYP7A1 die in the neonatal period, with severe malnourishment resulting from fat-soluble vitamin deficiencies and lipid malabsorption.4,11 The survival curve of animals surviving until day 18, however, subsequently flattens out, 11 and the observation that such animals are able to synthesize normal bile acids, in reduced number but sufficient for normal cholesterol metabolism, led to the description of the ‘alternate pathway’. 12
The alternate pathway uses oxysterols as starting substrates for bile acid synthesis rather than cholesterol, and primarily extra-hepatic. Sterol side chain oxidation is catalysed by a sterol hydroxylase (e.g. sterol 27 hydroxylase, CYP27A1), and then 7α hydroxylation is catalysed by 25-hydroxycholesterol 7α hydroxylase, CYP7B1. 4 The latter is the rate-limiting step. In healthy humans, these pathways are likely to contribute little to overall bile acid production, 13 but they play a particular role in cholesterol turnover in certain tissues including brain, vascular endothelium and lung.13–16
Oxysterol intermediates resulting from either pathway then undergo further modification in the liver. In the presence of sterol 12α-hydroxylase (CYP8B1), the intermediate is converted to cholic acid; in its absence, it is converted to chenodeoxycholic acid (CDCA) (rats, humans and hamsters) or β-muricholic acid (rodents). 4 Hepatic expression of CYP8B1 therefore regulates the ratio of cholic to CDCA, and the resulting hydrophobicity of the bile acid pool.4,17
Bile acid classification
Bile acids may be classified into primary, secondary and tertiary. Those synthesized from cholesterol in hepatocytes, as described in the previous section, are termed primary. 10
Primary bile acids are conjugated within hepatocytes before being secreted into bile. 10 There are five recognized types of conjugation, which occur at different locations of the steroid nucleus and side chain: N-acyl amidation with glycine or taurine; sulfation; ester glucuronidation; ethereal conjugation and N-acetylglucosamination. 6 The first of these, conjugation of the terminal side chain carboxylic acid with glycine or taurine, is performed by the enzyme bile acid CoA: amino acid N-acyltransferase (BAAT) and can be considered the final stage of bile acid synthesis, as it occurs to all newly synthesized bile acids in hepatocyte peroxisomes prior to excretion. 7
Conjugation with taurine or glycine increases bile acid solubility, and results in a molecule which is negatively charged at the pH of digestive fluids (pH 6–7). 6 Furthermore, the size of the conjugated molecule is too large to diffuse through paracellular junctions. Due to these factors, newly secreted conjugated bile acids are highly concentrated in the lumen of the small intestine, which is crucial to enable satisfactory digestion of lipids. 18
When primary bile acids are modified by enzymes, they become secondary bile acids; such modifications include removal, oxidation or epimerization of the nuclear hydroxyl groups. For example, the anaerobic bacteria in the caecum remove the hydroxyl group at C7 of CDCA and cholic acid, to form lithocholic acid (LCA) and deoxycholic acid (DCA), respectively. 6 Although some modifications are introduced by hepatic enzymes, gut bacteria play a major role in the metabolism of primary bile acids; indeed, germ-free rodents and those treated with antibiotics have far lower bile acid diversity than control animals.19,20 Bile acids which are then further biotransformed in the liver, after reabsorption from the gut, are in some texts named ‘tertiary’. 21
The complicated nomenclature of bile acids may be attributed to the fact that many were isolated long before their chemical structure was elucidated. For example, chenodeoxycholic acid was named after the species from which it was first isolated (‘chena’ means ‘goose’ in Greek), whereas deoxycholic acid was named for its differences to cholic acid, the first bile acid to be identified and assigned a chemical formula in the late 19th century. 22 In humans, the vast majority of circulating bile acids comprise cholic, DCA and CDCAs, and their glycine- and taurine-conjugated forms.23,24
Enterohepatic circulation of bile acids
There exists a highly efficient system for bile acid conservation and recycling within the body, which is termed the enterohepatic circulation. After synthesis in the liver, bile salts are secreted into bile ducts, from where they either enter the intestine or are stored in the gallbladder. 6 Upon meal ingestion, cholecystokinin drives gallbladder contraction and emptying, causing secretion of bile via the bile ducts into the intestine, where emulsification of nutrients occurs. 25 The vast majority of secreted bile acids (95%) then travel in the portal circulation back to the liver. 4 The remaining 5% is excreted in faeces and is replaced by newly synthesized bile acids in the liver from cholesterol. This cycle repeats 4 to 10 times daily. 26
Bile acid transport
As they move within the enterohepatic circulation, conjugated bile acids undergo active transportation from one compartment to another (reviewed by Zwicker and Agellon 10 ). The genes that express bile acid transporters are often regulated by bile acids themselves. This system allows for precise and responsive distribution of bile acids in accordance with meal status.
Major players in this process include the apical sodium-dependent bile acid transporter, expressed on the luminal side of ileal enterocytes, which is responsible for the uptake of luminal bile acids from the small bowel, 27 and the heterotrimeric organic solute transporter (OSTα/β) expressed on the basolateral membrane of the enterocytes, which secretes bile acids into the portal bloodstream. 28 In the liver, the sodium/taurocholate co-transporting polypeptide (NTCP) uptakes bile acids into hepatocytes. 29 These recycled bile acids, along with newly synthesized bile acids, are secreted into bile from the canalicular membrane of the hepatocytes via the bile salt export pump (BSEP or ABCB11).
Metabolic actions of bile acids (Table 1)
Major metabolic actions of bile acids (see text for details).
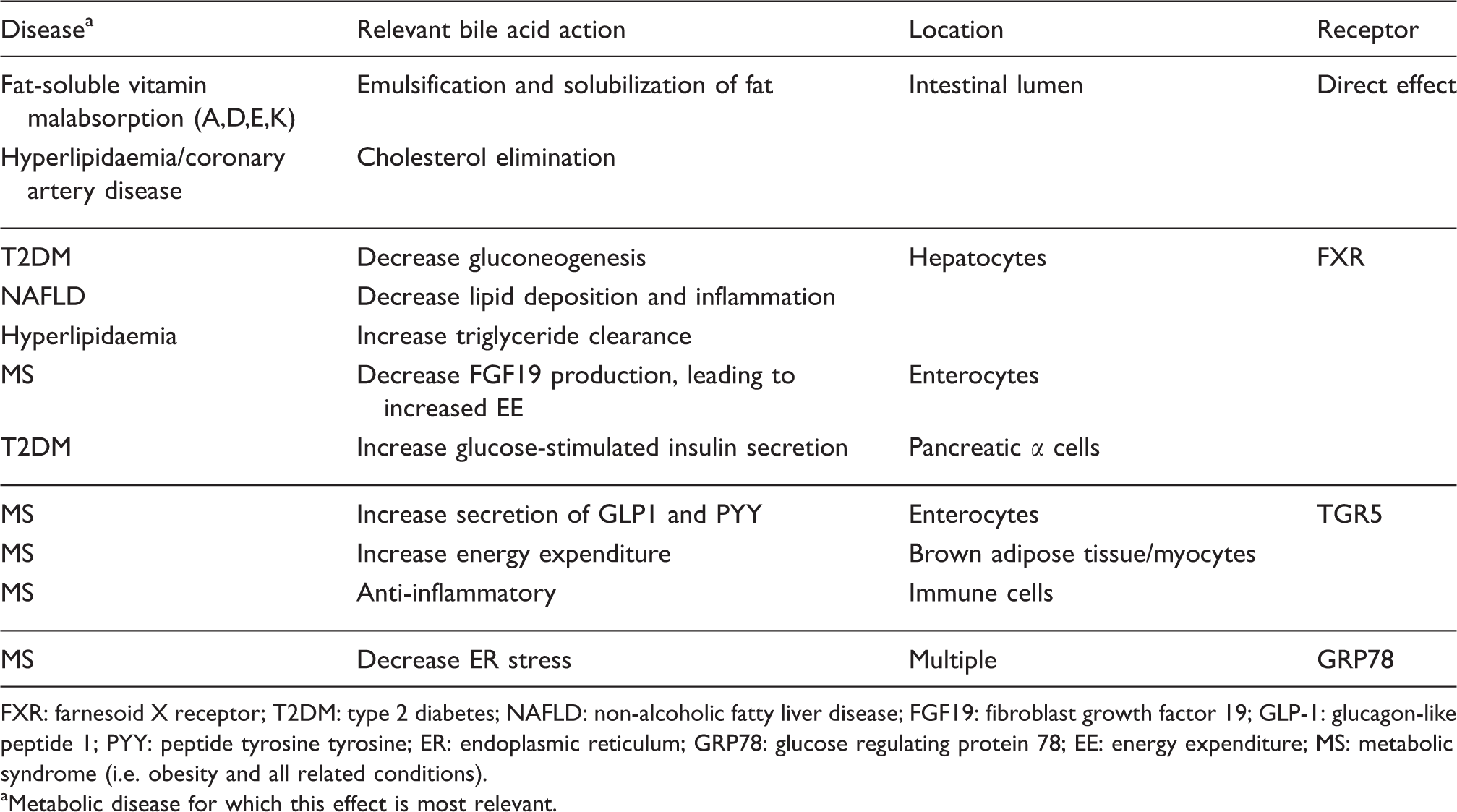
FXR: farnesoid X receptor; T2DM: type 2 diabetes; NAFLD: non-alcoholic fatty liver disease; FGF19: fibroblast growth factor 19; GLP-1: glucagon-like peptide 1; PYY: peptide tyrosine tyrosine; ER: endoplasmic reticulum; GRP78: glucose regulating protein 78; EE: energy expenditure; MS: metabolic syndrome (i.e. obesity and all related conditions).
aMetabolic disease for which this effect is most relevant.
Individual bile acids differ in their ability to bind to and stimulate different receptors. The great diversity of circulating bile acids, which are engaged in a dynamic process of synthesis, cycling and modification in response to environmental factors such as dietary intake, allows for tight modulation of bile acid-stimulated responses. 4
Lipid digestion and absorption
Bile acids emulsify dietary fats, converting them from fat globules into tiny droplets. The resulting increase in surface area renders the fats amenable to digestion by pancreatic enzymes. An example is bile salt-dependent lipase, which catalyses the hydrolysis of cholesterol esters into free cholesterol and fatty acids, enabling their absorption by the intestinal mucosa. 30 Bile acids also solubilize lipids by incorporating them into micelles. Certain essential vitamins (A, D, E and K) are non-polar lipids and require incorporation into micelles in order to be absorbed.31,32 In most animals and healthy humans, both the production of bile acids and their elimination are modulated in response to varying dietary intake of cholesterol.33,34 Diminished bile acid excretion predicts presence and severity of coronary artery disease.34,35 In this way, bile acid circulation plays a key role in physiological cholesterol homeostasis. 36
Actions of bile acids mediated by FXR
FXR is a nuclear transcription factor, which forms a complex with retinoic X receptor and then binds to gene promoter regions. 37 FXR is highly expressed in human liver and gut, as well as in kidney, adrenals, vascular smooth muscle, white adipose tissue and immune cells. 38 Various biological features regulate and integrate the activity of FXR with other stimuli. Firstly, there are four isoforms of FXR, which are differentially expressed in different tissues. 39 The isoforms vary in their responsiveness to bile acid stimulation, with the isoform most commonly expressed in liver (FXRα1) exhibiting the greatest responsiveness in vitro. 40 Secondly, as a typical eukaryote nuclear transcription factor, activation of FXR releases corepressor complexes and/or stabilizes coactivator complexes in the promoter region of target genes. Multiple cofactors interact with FXR, and their recruitment is often also ligand dependent. 38
FXR is critical to the homeostatic-negative feedback cycle, which regulates bile acid synthesis and distribution; and additionally, it has key beneficial metabolic effects on multiple systems.
In hepatocytes, FXR activation increases small heterodimer partner (SHP) expression, which represses CYP7A1 expression, thereby reducing synthesis of bile acids via the classic pathway. 41 SHP also represses NTCP, which decreases uptake of bile acids by the liver from the portal circulation. 42 FXR increases bile acid efflux from the liver into bile by increasing BSEP expression 43 and also into the portal circulation by upregulating OSTα/β. 44 In these ways, hepatocyte FXR detects intracellular bile acids and activates mechanisms to decrease their excessive and potentially toxic accumulation.
Additionally, the activation of SHP suppresses CYP8B1 activity, which decreases the ratio of cholic acid to CDCA. A relative decrease in cholate to other bile acids increases the hydrophilicity of the circulating bile acid pool, thereby decreasing intestinal cholesterol absorption.4,45 Cholic acid itself is a negative regulator of CYP7A1, and reduction in circulating cholic acid increases CYP7A1 activation. 46 Thus, a negative feedback loop maintains bile acid production in this complex system.
In human enterocytes, FXR activation increases production and secretion of fibroblast growth factor 19 (FGF19; or its orthologue FGF15, in mice). This circulates in portal blood and activates the hepatic fibroblast growth factor receptor 4 (FGFR4). Activation of FGFR4 also powerfully represses CYP7A1. Although FGF19 is also released from hepatocytes on FXR activation, 47 the intestinal route is likely to be the dominant effector of CYP7A1 repression. 48 In obese rodents, FGF19 has been shown to act centrally to increase energy expenditure, decrease weight and improve glucose tolerance. 49
FXR increases hepatic triglyceride clearance, by modifying the expression of several genes involved in lipid transport and metabolism. For example, it reduces the expression of apolipoprotein CIII 50 and increases the expression of apolipoprotein CII, 51 apolipoprotein E 52 and phospholipid transfer protein. 53 Furthermore, FXR increases hepatic uptake of low-density lipoprotein (LDL) and very low density lipoprotein (VLDL) by increasing hepatocyte expression of syndecan-1, a transmembrane proteoglycan that participates in LDL binding and internalization, and VLDL-receptor.54,55 Accordingly, FXR(–/–) mice have increased plasma cholesterol and triglycerides. 56
Deletion of FXR increases susceptibility to hepatic steatosis in mice.57,58 FXR activation may be directly protective for NAFLD, by decreasing hepatic lipid deposition and activating anti-inflammatory pathways. 59 Patients with non-alcoholic steatohepatitis (NASH) have lower hepatic protein expression of FXR and SHP compared to those with simple steatosis. 60 Key nuclear transcription factors involved in hepatic lipid metabolism include sterol regulatory element-binding protein 1c, which promotes lipogenesis and is repressed by FXR, and peroxisome proliferator-activated receptor α, which increases fatty acid oxidation and is increased by FXR. 61 A major mechanism through which FXR decreases inflammation in the liver is through nuclear factor κβ, a process which can be reversed by aberrant acetylation of FXR. 62
Hepatic FXR also governs hepatic gluconeogenesis. Via SHP, it suppresses key enzymes involved in this pathway, including phosphoenol-pyruvate carboxykinase and glucose 6-phosphatase (G6Pase). 58 In adipocytes, both FXR agonism and stimulation with FGF19 cause increased glucose uptake.63,64 Furthermore, in pancreatic β cells, FXR agonism inhibits membrane potassium channels, which increases calcium influx, resulting in greater glucose-stimulated insulin secretion.65,66
Actions mediated by TGR5
TGR5 is a bile acid receptor located on the cell membrane, 67 also known as the membrane-bile acid receptor or G protein bile acid-activated receptor. 68 Gene expression of this receptor is ubiquitous in human and rodent tissues, with higher levels in lung, liver, gallbladder, spleen, fat, central nervous system and gut. 68 Immunohistochemical analysis of mouse gastrointestinal tract has shown that TGR5 is found in the enteric nervous system, mainly in inhibitory neurons. 69 TGR5 is a proto-typical transmembrane G protein-coupled receptor, activation of which leads to intracellular signalling via adenylyl cyclase and cyclic AMP. 67 Depending on the cell line, TGR5 activation may also lead to increase or lowering of intracellular calcium levels.70,71 In vitro, LCA, DCA and CDCA stimulate TGR5 robustly, whereas ursodeoxycholic acid (UDCA) does not. 72
Within the liver, TGR5 is abundant in non-hepatocytes, including cholangiocytes, gallbladder epithelial cells, Kupffer immune cells and liver sinusoidal epithelium. Stimulation of TGR5 in these biliary tree locations allows bile acids to act in a paracrine manner, streamlining biliary activity according to bile acid flux. For example, activation of TGR5 in vascular endothelial cells causes vasodilation, increasing hepatic blood flow. 73 TGR5 activation in gallbladder epithelium leads to chloride secretion into bile, probably via the cystic fibrosis transmembrane chloride channel. 74 TGR5 stimulation also relaxes the smooth muscle of the gallbladder wall, allowing it to fill with bile. 75
Bile acids have also been known for some time to stimulate enterocyte secretion of gut hormones involved in blood sugar and appetite regulation. 76 These include glucagon-like peptide 1, an incretin hormone which augments insulin secretion in response to oral glucose; and peptide YY, a centrally active anorectic hormone. Recent mechanistic studies demonstrate that these effects are likely to occur secondary to intestinal absorption of bile acids, primarily via stimulation of TGR5 on the basolateral membranes of enterocytes.72,77
Another key beneficial metabolic effect of bile acids occurs via the TGR5 receptor in brown adipose tissue (BAT). In mice, bile acid activation of TGR5 in BAT increases energy expenditure, thereby providing resistance to obesity and improvements in metabolic health. 78 This occurs via activation of type 2 iodothyronine deiodinase, which converts inactive thyroxine into active 3,5,3′-triiodothyronine. Although adult humans have only small amounts of BAT, the same pathway has been shown to occur in vitro in human skeletal myocytes, provoking increased cellular energy expenditure. 78
Other biological effects of bile acids mediated through TGR5 include anti-inflammatory effects in monocytes and macrophages.79,80 Stimulation of TGR5 inhibits production of pro-inflammatory cytokines. TGR5 mediates the stimulatory effects of bile acids on colonic peristalsis, and lack of this pathway results in constipation in mice. 81 Additionally, TGR5 on sensory nerves mediates pruritus and analgesia, 82 characteristic symptoms of patients with obstructing biliary disease and hence high circulating bile acids. 83
Reduction of endoplasmic stress
The endoplasmic reticulum (ER) is responsible for folding new proteins via chaperones and foldases. Where the ER’s capacity for protein folding is overwhelmed (for example, when there is excessive protein synthesis or abnormal proteins), misfolded proteins accumulate in the ER and cause ER stress. 84 In response, the unfolded protein response (UPR) is triggered. Initially, this is adaptive, preventing translation of further proteins and promoting autophagy of existing proteins, but if ER homeostasis is not regained, it ultimately leads to cell apoptosis.
ER stress is increased in obesity, 85 and it is heavily implicated in the pathogenesis of obesity and diseases of insulin resistance. In adipose tissues, ER stress induces inflammation and the secretion of harmful adipokines; in pancreatic tissues, ER stress can impair insulin synthesis. 86 Additionally, ER stress leads to phosphorylation of insulin receptor-substrate 1 and suppression of insulin receptor signalling in liver and adipose tissues. 87
Tauroursodeoxycholate (TUDCA) has been demonstrated in vitro to decrease ER stress in hepatoma cells, intestinal epithelial cells and cardiac myoblasts.87,88 Cholic acid, a major endogenous bile acid, also reduces tunicamycin-induced ER stress in cultured hepatoma cells, adipocytes and pancreatic β-cells. 89 The mechanism is not fully understood but appears to involve the modulation of expression of a key ER chaperone glucose regulating protein, GRP78. GRP78 is increased via TUDCA-mediated phosphorylation of upstream molecules (PI3K/Akt and PKG) in cardiac cells 88 and decreased by UDCA in intestinal cells, in both cases reducing ER stress.
Changes in bile acid circulation in health and disease
Total and individual bile acid concentrations in different physiological compartments are highly variable with metabolic state. The chemical diversity of different bile acid species and their wide concentration ranges can make comprehensive analysis challenging. There is often a compromise between bile acid coverage and rate of throughput. 91 Bile acids are most frequently analysed from plasma of peripheral blood, although they can be extracted from other biofluids, commonly faeces, luminal gut content and urine. 92
Circulating bile acid fluctuations in metabolic health
Postprandially, there is an increase in delivery of bile acids to the duodenum, from where they are efficiently returned via the portal circulation to hepatocytes. Postprandial concentrations of portal vein bile acids increase up to six-fold, peaking at 15–60 min after a meal. 93 Hepatocytes clear bile acids into bile canaliculi and systemic blood; the rate of the former remains relatively constant, while the spillover rate into circulating blood is adjusted according to the rate of delivery to hepatocytes.93,94 There is a good correlation between portal and peripheral venous bile acid concentrations, with the former approximately six to seven times higher than the latter. 93 Fasting total serum bile acids are typically below 5 μM, and may rise as high as 10–15 μM postprandially.23,94,95 Meals with a higher fat content stimulate a higher postprandial bile acid rise. 23 Peak circulating bile acids are seen at around 1 to 2 hours postprandially.23,96
Hepatic fractional uptake varies between bile acid species, such that there is an enrichment of systemic glycine-conjugated primary bile acids postprandially.23,96 A similar response is seen after ingestion of glucose alone, with a rise in conjugated bile acids from 30 min after oral glucose tolerance test (OGTT).97–99
Fasting total plasma bile acid concentrations have been reported to be higher in men than women, 100 although a study of non-fasting samples found them to be non-significantly higher in women. 24 One study reported that concentrations of most individual fasting bile acids falls with age, although for several species of bile acids this was gender dependent 100 ; conversely, another group demonstrated no change with age. 24 There may also be ethnic differences in circulating bile acids, with a recent report of higher serum concentrations of glycocholic acid, glycochenodeoxycholic acid (GCDCA), CDCA and taurochenodeoxycholic acid in Asians compared with Caucasians. 24 The variation in study conclusions may well relate to unexplored biological variables as well as assay variability.
Fluctuations in bile acid synthesis
As outlined earlier, de novo synthesis of bile acids is regulated by negative feedback mechanisms; bile acid-stimulated secretion of FGF19 from enterocytes plays a particularly important role by suppressing hepatocyte CYP7A1. Administration of CDCA orally for two weeks increases circulating FGF19, decreases CYP7A1 activity and decreases endogenous bile acid synthesis.101,102 Conversely, treatment with cholestyramine, a bile acid sequestrant which prevents intestinal bile acid resorption, leads to a decrease in circulating FGF19, increase in hepatic CYP7A1 activity and increased bile acid synthesis.101,102
Bile acid synthesis has a diurnal pattern, with peaks occurring in the middle of the day and in the evening. 103 Rate of synthesis is higher in men than in women and positively correlated with plasma triglyceride level. 26 Bile acid synthesis decreases with age, and this is associated with an increase in plasma cholesterol. 104 An increased rate of bile acid synthesis is associated with the development of gallstone disease. 105 An increased rate has also been demonstrated in volunteers lacking a gallbladder. 26
Bile acid homeostasis in obesity and diseases of insulin resistance
To date, studies of bile acid pool size have involved small numbers of individuals, and results are often contradictory. Most agree that mildly insulin-resistant individuals tend towards higher total fasting circulating bile acids.106–109 Some23,107–109 but not all 95 studies reported higher fasting total bile acids in patients with T2DM. Similarly, there is controversy regarding the level of total bile acids in fasting obese patients: two studies reported comparable levels to normal weight controls,96,110 while another reported higher total bile acids in obese patients. 106
In patients with obesity, the rise in circulating bile acids in response to a meal stimulus is suppressed.96,110 Metabolomic profiling of patients with pre-diabetes demonstrated that the increase in GCDCA during an OGTT is reduced, and negatively correlates with fasting insulin. 98 In contrast, Vincent et al. demonstrated that patients with obesity and T2DM experience a higher peak change in total bile acids and in total glycine-conjugated bile acids compared with matched normoglycaemic controls when challenged with a standard meal. 111 Similarly, a recent study looking at OGTT and meals with differential fat content reported that in all but the high-fat meal, patients with T2DM experienced a higher area under the curve for postprandial total bile acids than the matched controls. 23
In terms of composition of the bile acid pool, obesity has been associated with a higher cholic acid content of the bile pool. 106 Similarly, insulin resistance has been associated with an increased fasting ratio of 12α-hydroxylated bile acids (cholic acid, DCA and derivatives) to non-12α hydroxylated forms (CDCA, LCA and other derivatives).95,108 This has been attributed to a reduction in insulin stimulation of hepatic forkhead box protein O1, which ordinarily suppresses CYP8B1 and therefore decreases conversion of bile acid intermediates to cholic acid. 112 A recent study of patients with T2DM demonstrated that most fasting bile acid species were higher than in matched controls, and that postprandially patients with T2DM had higher 12α-hydroxylated bile acids with comparable CDCA and its derivatives. 23 Wewalka et al. reported a correlation between total taurine-conjugated species and insulin resistance, 107 with a trend towards higher fasting taurine–cholic acid but not glycine–cholic acid in patients with T2DM.
Plasma metabolomic profiling of non-diabetic patients with histologically confirmed non-alcoholic fatty liver compared with healthy age- and sex-matched controls revealed significantly higher fasting concentrations of conjugated bile acids. 113 Similarly, higher fasting and postprandial excursions of circulating total and conjugated bile acids were found in a small study of patients with confirmed NASH. 114 Potential confounders of both studies include the fact that although all volunteers were non-diabetic, patients with steatosis had higher BMI and higher indices of insulin resistance than controls. A further study of patients with liver impairments from all causes also demonstrated higher total and conjugated serum bile acids, 24 suggesting that these changes are common to all liver injury rather than specific to NAFL and insulin resistance.
As NAFL progresses to NASH and then cirrhosis, there is evidence that bile acid synthesis gene expression changes, with upregulation of CYP7B1 and thus promotion of the alternative pathway of bile acid synthesis. 115 BAAT expression and taurine availability increase, resulting in a change in composition of hepatocyte bile acids towards taurine-conjugated species. 115 Total hepatic bile acid content is higher in patients with steatohepatitis than in matched controls, whether of alcoholic or non-alcoholic origin. 116
Contribution of the gut microbiome
The gut microbiota, which is an extraordinarily diverse and plastic community of symbiotic bacterial species, is responsible for biotransformation of primary to secondary bile acids as they pass through the gut. As bacterial enzymes that conjugate bile acids vary according to bacterial strain, the microbial community is a key determinant of bile acid pool size and composition.44,117 Oral agents that alter the microbiome, such as probiotics, antibiotics and antioxidants, change bile acid profiles: mechanisms include changes in intestinal conjugation of bile acids and differential expression of hepatic and intestinal bile acid transporters.118–120 Furthermore, bile acids themselves affect the composition of the gut microbiome, via direct detergent effects on bacteria as well as effects on intestinal mucosa integrity. 121
Microbiome changes are well documented in association with NAFLD,122,123 obesity and T2DM. 124 The mechanisms by which the microbiome influences metabolic health extend beyond its effects on bile acid transformation. For example, different bacterial communities produce different metabolites, which act as local signalling molecules with differential effects on mucosal inflammation, intestinal permeability and gut hormone production. 125 The relationships between metabolic disease, bile acids and the gut microbiota are therefore complex and interdependent.
Bile acid manipulation for metabolic diseases
Given the potentially beneficial metabolic actions of bile acids, there has been much interest in harnessing their effects as therapy for metabolic syndrome diseases. Bile acids, bile acid-binding resins and synthetic FXR and TGR5 agonists have been investigated for this purpose. Due to the complex negative feedback cycles involved in bile acid homeostasis, the use of these types of agents is not straightforward, as they may have variable and unpredictable effects on overall bile acid pool size and composition. 66 In part, the effect of treatment on circulating bile acids depends on pretreatment bile acid pool, which varies with the presence and/or severity of metabolic disease.
Bile acid-binding sequestrants
Bile acid-binding resins or sequestrants (BAS) are non-absorbable cationic polymers that bind bile acids in the gut and prevent their resorption. Use of these agents, which include colesevelam, cholestyramine and sevelamer, reduces the return of intestinal bile acids to the liver, thereby stimulating the synthesis of new bile acids from cholesterol. 126 Used as single agents or as part of combination therapy, BAS are well known to reduce lipidaemia and decrease coronary artery disease risk. 127
Colesevelam as an adjunct to other lipid-lowering therapies in patients with poorly controlled T2DM has been shown in several randomized controlled trials (RCTs) to reduce glycaemia and lipidaemia.126,128 Colestimide as an add-on to lifestyle modification has been shown in an RCT to confer additional benefits for patients with NASH: better improvement in liver histology and transaminases, greater decrease in visceral fat and enhanced improvement in glycated haemoglobin (HbA1c). 129 Sevelamer has been shown in a mouse model of NAFLD to improve hepatic steatosis and inflammation in NASH by inhibiting FXR signalling 130 ; human trials are awaited. Colesevelam, however, has not been shown to confer the same benefits and has been associated with an increase in liver fat in both mice and patients with biopsy-proven NASH.131,132
The exact mechanism of the positive metabolic effects of BAS is incompletely understood.126,133 Up to eight weeks of treatment with colesevelam does not increase total bile acid pool size in either healthy volunteers or patients with T2DM, but doubles the cholic acid proportion of the pool. 95 Colesevelam treatment has also been shown to increase fasting and postprandial levels of incretin hormones glucagon-like peptide 1 and gastric inhibitory polypeptide. 134 The mechanism for this is likely due to the fact that BAS retain bile acids in the gut, allowing them to stimulate TGR5 receptor in the colon (in normal physiology, almost all bile acids are reabsorbed in the small intestine, with very few reaching the colon). 135 Differential binding affinities for different bile acid species may explain differences in clinical effect conferred by use of different BAS. 126
Bile acids
UDCA is used for the treatment of primary biliary cirrhosis, and in this context has been shown to activate anti-inflammatory hepatocyte pathways and improve liver function. 136 As a treatment for NASH, however, there have been several RCTs 137 with results that are insufficient for its recommendation by any of the major practice guidelines. 138 It is worth noting that UDCA is inactive at the FXR receptor at physiological concentrations. 38 CDCA has recently been demonstrated in mice on a high-fat diet (HFD) to decrease weight and improve glucose tolerance. 139
Synthetic drugs that target bile acid receptors
Due to the regulatory role of FXR stimulation on glucose and lipid metabolism, multiple FXR-specific agonists are in development. 140 CDCA is the most potent natural ligand for FXR 38 ; therefore, it has been used as the starting point for the development of synthetic FXR agonists. Obeticholic acid (OCA; INT747) is the best characterized of these. It is a synthetic 6α ethyl derivative of CDCA and a highly potent FXR agonist, and is administered as an oral tablet.
In a multicentre randomized control trial, patients with histologically confirmed non-cirrhotic NASH were treated with OCA or placebo for 72 weeks. 141 Patients treated with OCA had significantly greater improvements in liver histology, including better reduction in steatosis, inflammation and fibrosis. They also had an approximate 2 kg reduction in weight and decreases in alanine aminotransferase and gamma glutamyltranspetidase, but an increase in aspartate aminotransferase. Patients treated with OCA experienced a rise in serum cholesterol and insulin resistance (as measured by homeostatic model assessment (HOMA)); they also experienced a higher incidence of pruritus than placebo. 141 Clinical findings were similar to those of a shorter six-week study of patients with T2DM and NAFLD; however in this latter study, which used hyperinsulinaemic euglycaemic clamps, insulin sensitivity improved with OCA treatment. 142 The finding of increased serum cholesterol is predictable, due to the suppression of bile acid synthesis by FXR activity; it is unclear at this stage if this effect is linked to adverse clinical outcomes. Pruritus may be due to cross-reactivity with the TGR5 receptor. 82 The results of a phase III study of OCA in NASH with fibrosis are awaited. 140
A potential problem with systemic FXR agonism is that the effects appear to be strikingly different depending on the animal model studied. For example, the synthetic FXR agonist GW4064 improves glucose tolerance in wild-type and db/db mice, 143 but exacerbates weight gain and glucose intolerance, and decreases energy expenditure, in mice on a HFD. 144 This might be explained by different hepatic transcription pathways secondary to FXR stimulation in obese mice when compared with lean. 145 Furthermore, administration of GW4064 dramatically changes the bile acid pool size and composition 144 ; different starting bile acid pool size and composition in animals with metabolic disease compared with healthy counterparts may be altered in different ways.
One response to this issue is the development of non-steroidal FXR agonists. In contrast to OCA and other bile acid-derived compounds, an advantage of these is that they are expected not to undergo enterohepatic circulation and therefore have more predictable pharmacokinetics. GS-9674 and LJN452 are two such compounds currently in phase II clinical trials. 140
Another approach is an FXR intestine-specific agonist, for example fexaramine, which is a poorly absorbed oral agent and is therefore intestinally restricted. This has been found to reduce weight gain and improve glucose tolerance in diet-induced obese (DIO) mice. 146 Paradoxically, an intestinal FXR antagonist, glycine-β-MCA, has also been shown to improve metabolic phenotype of mice on a HFD. 147
TGR5 agonism is also beneficial for metabolic disease, primarily by increasing energy expenditure. The TGR5-specific agonist INT777 decreases weight in DIO mice, increases energy expenditure and decreases hepatic steatosis.148,149
Finally, some compounds in development are dual agonists at both the FXR and TGR5 receptor. One such is INT767, which has been shown to improve steatohepatitis in obese diabetic mice over six weeks of treatment, despite no significant change in body weight or glucose tolerance. 150
Conclusion
Given the global pandemic of obesity and related disorders, and the escalating understanding of bile acids as powerful regulators of metabolism, this is a very exciting time for bile acid biology. Generally, the actions of bile acids are positive for metabolic health and include elimination of dietary cholesterol, stimulation of gut hormone release, increased energy expenditure, reduction of liver fat and inflammation and reduction of ER stress. Circulating bile acid levels are highly responsive to physiological and pathophysiological variables, which can make their assessment difficult. Nonetheless, most evidence suggests that both fasting and postprandial circulating levels are higher in metabolic disease. This could potentially represent a compensatory state where an increase in bile acids is a physiological response to alleviate further development of metabolic disease.
In terms of pharmacological use of bile acids and related compounds as therapy for metabolic diseases, the field is highly active with many new drugs being synthesized and trialled. One challenge is the tight homeostatic response to bile acid manipulation, which means that overall effect on pool size and composition of bile acids reaching target tissues is often difficult to predict. Furthermore, the activity of major bile acid receptor FXR appears to differ in different disease states, affecting the response to bile acid manipulation. Nonetheless, the powerful metabolic actions of bile acids and their generally good safety profile make them excellent therapeutic targets. Further developments in this field are eagerly anticipated.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The Section of Endocrinology and Investigative Medicine is funded by grants from the MRC, BBSRC, NIHR, an Integrative Mammalian Biology (IMB) Capacity Building Award, an FP7- HEALTH-2009– 241592 EuroCHIP grant and is supported by the NIHR Biomedical Research Centre Funding Scheme. The views expressed are those of the authors and not necessarily those of aforementioned funders, the NHS, the NIHR or the Department of Health. ER McGlone is also funded by the Medical Research Council with a clinical research training fellowship.
Ethical approval
Not applicable.
Guarantor
ERM.
Contributorship
Both authors conceived the topic. ERM wrote the first draft of the article, which was reviewed by SRB. Both authors approved the final version of the article.