
Abstract
Background
Measurement of serum dexamethasone during the overnight dexamethasone-suppression test has been recommended to reduce false-positive results when investigating Cushing’s syndrome or increasingly commonly found adrenal incidentalomas. Despite this, there remains a paucity of well-validated dexamethasone methods currently available. Here, we describe the development of a rapid and sensitive liquid chromatography tandem mass spectrometry serum dexamethasone assay and verify its utility in a cohort of postmenopausal females.
Method
Isotopically labelled internal standard was added to samples prior to supported liquid extraction. Extracts were analysed using liquid chromatography tandem mass spectrometry in the positive electrospray ionization mode. Multiple reaction monitoring was used to detect dexamethasone and its corresponding internal standard transitions. Normal healthy postmenopausal women (n = 95) were recruited and underwent an overnight dexamethasone suppression test, with serum dexamethasone and cortisol measurements at 09:00 after administration of oral dexamethasone 1 mg at 23:00 the night before.
Results
Mean intra- and inter-assay imprecision were 4.1% and 2.9%, respectively, for dexamethasone concentrations of 1.5, 6.0 and 12.0 nmol/L. Matrix effects were found to be negligible at 106–109% with recovery ranging from 96 to 100%. The limit of quantitation was 0.25 nmol/L, and structural analogue analysis proved the method to be robust against interferences. Applying a serum dexamethasone cut-off of >3.3 nmol/L was associated with a serum cortisol ≤50 nmol/L in 84/95 individuals.
Conclusion
We have developed a sensitive and robust liquid chromatography tandem mass spectrometry method for the quantitation of serum dexamethasone. The method can be used to identify false-positive results during the overnight dexamethasone suppression test or for pharmacokinetic studies.
Introduction
Dexamethasone is a potent synthetic fluorinated glucocorticoid widely used for the treatment of inflammatory and autoimmune conditions. In addition to its therapeutic applications, its high affinity for the glucocorticoid receptor and low cross-reactivity with routine cortisol assays have helped establish a diagnostic application. Here, its administration serves to challenge the hypothalamic-pituitary-adrenal axis to temporarily suppress cortisol production. This property has been utilized to develop a screening test for the investigation of hypercortisolism in patients suspected of having Cushing’s syndrome (CS) 1 or autonomous cortisol secretion in patients with an adrenal incidentaloma (AI). 2
The protocol for the overnight dexamethasone suppression test (ONDST) is now well established and involves the administration of 1 mg oral dexamethasone at 23:00 followed by the measurement of serum cortisol at 09:00. 1 An 09:00 serum cortisol concentration ≤50 nmol/L is deemed a normal response and is sensitive (>95%) and specific (80%) for the exclusion of hypercortisolism.1,2 Patients who do not adequately suppress cortisol warrant further investigation in the form of additional biochemical testing or imaging.1,2
Despite the high sensitivity of the ONDST, it is widely recognized that false-positives may be observed due to several conditions including: poor intestinal absorption of dexamethasone, variation in cortisol-binding globulin concentrations, impaired hepatic or renal clearance of dexamethasone, drug induction or inactivation of the hepatic CYP3A4 enzyme pathway or simply interindividual variation in the of metabolism dexamethasone. 1
Thus, to reduce false-positive results, it has been recommended that quantitation of serum dexamethasone alongside serum cortisol may help to improve the diagnostic accuracy of the ONDST, thereby limiting the unnecessary and often expensive over-investigation of patients. 1 Indeed, there is now growing evidence to support this and post-ONDST dexamethasone concentrations >5.6 nmol/L 3 and >3.3 nmol/L 4 have been proposed.
Despite this, there is a paucity of well-validated dexamethasone methods available. Early methods for dexamethasone relied on radioimmunoassay (RIA) and utilized a highly specific antibody to differentiate dexamethasone from structurally similar compounds. 3 More recently, several liquid chromatography tandem mass spectrometry (LC-MS/MS) methods have been published. Many of these rely on solid phase extraction (SPE) for sample preparation,5–8 protracted run-times, 9 or are not sensitive enough to be used in combination with the ONDST.10,11 These issues have limited the widespread application of serum dexamethasone measurement into the workflow of busy, high-throughput clinical laboratories.
Here, we sought to develop a rapid and sensitive LC-MS/MS method for serum dexamethasone quantitation that could be used for routine clinical use. In addition, we looked to verify the proposed >3.3 nmol/L dexamethasone cut-off.
Materials and methods
Different lot numbers of dexamethasone certified reference material (CRM) were used at a concentration of 1 mg/mL as stock solutions for calibrators and quality control (QC) materials (Cerilliant, Texas, USA). Stable deuterium-labelled dexamethasone-d4 (4, 6α, 21, 21) with an isotopic purity >98% (CDN isotopes, Quebec, Canada) was used as an internal standard at a working concentration of 120 nmol/L in methanol (LC-MS grade, Honeywell, New Jersey, USA).
Preparation of calibrators and QC materials
Dexamethasone calibrators and QC materials were prepared in phosphate-buffered saline (PBS) 0.1% (wt/vol) bovine serum albumin (BSA, Sigma, Dorset, UK). Calibrators ranged from 0 to 16 nmol/L (n = 7) and QC materials were prepared at concentrations of 1.5, 6.0 and 12.0 nmol/L.
Preparation of samples
Each calibrator, QC material and sample was manually pipetted using a positive displacement pipette (Gilson Microman®, Middleton, UK) in 100 μL aliquots into the wells of a 96-well plate (Porvair Sciences Ltd, Leatherhead, UK). To this, internal standard (10 μL) and deionized water (100 μL) were added to each well before the plate was heat sealed and vortex mixed for 1 min (Grant Instruments MPS-1, Cambridge, UK). The entire contents of each well were then transferred to the corresponding well of a 200 μL supported liquid extraction (SLE) plate (Biotage®, Uppsala, Sweden), drawn on to the diatomaceous earth using a vacuum pump for 30 s (KNF Neuberger, Oxford, UK) and left for 20 min. Dexamethasone was subsequently eluted from the SLE plate using, 1000 μL methyl tert-butyl ether (HPLC grade, Fisher Scientific, Loughborough, UK) and collected into a second 96-well plate. The resulting eluate was dried under air (Biotage® SPE Dry, Biotage®, Uppsala, Sweden), reconstituted in 100 μL of 40% methanol/water (vol/vol), heat sealed, vortex mixed for 1 min and centrifuged at 880 × g for 5 min.
Chromatography
We used an Acquity® ultra-performance liquid chromatography I-Class system (Waters, Manchester, UK) to achieve chromatographic separation. Mobile phases consisted of (A) deionized water with 2 mmol/L (154.2 mg/L) ammonium acetate (LC-MS grade, Sigma, Poole, UK) and 0.1% (vol/vol) formic acid (VWR International, Lutterworth, Leicestershire) and (B) methanol with 2 mmol/L (154.2 mg/L) ammonium acetate and 0.1% (vol/vol) formic acid. The prepared sample (15 μL) was injected onto a KrudKatcher® Ultra HPLC 0.5-μm in-line filter (Phenomenex, Macclesfield, UK) connected in series to a C8 2.6 μm, 30 × 2.1 mm analytical column (Phenomenex, Macclesfield, UK). Starting conditions were 40% mobile phase B at a flow rate of 0.6 mL/min, this was increased linearly over 1.1 min to 55% B. After this, the mobile phase composition was increased to 98% B and held for 30 s before returning to 40% B to re-equilibrate. Total run time injection-to-injection was 2.2 min. The auto-sampler was held at 10°C and the column oven temperature was maintained at 35°C throughout.
Mass spectrometry
Following chromatographic separation, the eluate was directed into a Waters Acquity Xevo TQS Micro® tandem mass spectrometer with a Z-spray ion source (Waters, Manchester, UK). We used MassLynx version 4.1 software for system control and MassLynx TargetLynx for data processing. The mass spectrometer was operated in the positive electrospray ionization mode, the capillary voltage was maintained at 0.4 kV and the source temperature was 150°C. The desolvation temperature and gas flow were 500°C and 900 L/h, respectively. For dexamethasone, the m/z 393 > 237 (quantifying) and m/z 393 > 147 (confirmatory) transitions were monitored; for dexamethasone-d4, the corresponding m/z 397 > 239 transition was monitored. The cone and collision energies were 35 V and 18 V, respectively, for both the quantifying ion (QI) and dexathasone-d4 transitions. The confirmatory ion (CI) had a cone energy of 35 V and a collision energy of 28 V. All transitions were monitored in the multiple reaction monitoring (MRM) mode with a dwell time of 0.088 s.
Validation
The assay performance characteristics were validated against the Food and Drug Administration’s (FDA) published criteria.12,13 Unless stated otherwise, all validation were undertaken using anonymized, surplus serum collected in 5 mL serum separator tubes (BD, Wokingham, UK).
Accuracy
In accordance with the FDA, accuracy has been assessed through analysis of gravimetrically prepared PBS 0.1% BSA QC materials at three concentrations over 10 consecutive batches.12,13 At each concentration, the mean deviation from the true concentration should not exceed 15%.12,13 The method of standard additions was used with spiked serum (n = 5) and PBS 0.1% BSA samples to confirm the use of the latter as a suitable matrix for calibrators and QCs. Agreement between the two matrices within 5% of the target concentration was deemed acceptable.
Imprecision
The imprecision of the method was assessed using QC materials. To determine the intra-assay imprecision, the three levels of QC materials were analysed 10 times within one batch. To investigate the inter-assay imprecision, the same QC materials were analysed daily over 10 days. At each concentration, the imprecision should not exceed 15% of the coefficient of variation (CV).12,13
Recovery
Serum samples (n = 5) were spiked with six different concentrations of dexamethasone CRM ranging from 0.5 to 16 nmol/L. Recovery was calculated using (measured−target/target × 100). We deemed recovery between 80 and 120% acceptable.
Matrix effect, extraction efficiency and process efficiency
Matrix effects (i.e. ion suppression/enhancement), extraction efficiency and the process efficiency were assessed in accordance with Matuszewski et al. 14 To quantitate the matrix effect, serum samples (n = 5) were spiked postextraction with seven different concentrations of dexamethasone ranging from 0 to 32 nmol/L. The analyte to internal standard peak height responses were then compared with their corresponding responses in un-extracted dexamethasone solvent standards in 40% methanol/water (vol/vol). To assess the extraction efficiency, the same serum samples were spiked pre-extraction using the same concentrations as detailed above. The pre-extracted analyte to internal standard peak height responses were then compared with their corresponding postextract responses. The overall process efficiency was calculated from the product of the matrix effect and extraction efficiency divided by 100. For each of these criteria, we defined acceptability as 80–120%.
Limit of quantitation
The limit of quantitation (LOQ) was defined as the concentration for which 10 replicates of serum samples prepared with low concentrations of dexamethasone produced a CV <20% and bias no greater than 20% while maintaining a signal-to-noise ratio of 5:1.12,13
Linearity
To assess linearity, we prepared 10 calibration curves in separate batches. The ratio of analyte peak height to internal standard peak height was plotted against dexamethasone concentration (nmol/L). A 1/X weighted linear regression model was fitted to the individual data-sets with calibration curves judged linear if the correlation coefficient (R2) was ≥ 0.99.
Linearity on dilution
To investigate the suitability of PBS 0.1% BSA as a sample diluent and to confirm a linear relationship between the analyte concentration and response, dexamethasone-free serum (n = 5) was spiked with dexamethasone to a final concentration of 16 nmol/L. The samples were then diluted with PBS 0.1% BSA to target concentrations of 1, 2, 4, 8 and 12 nmol/L and assayed as described above. The measured concentration was then multiplied by the appropriate dilution factor and the deviation from the original concentration calculated. For all dilutions, we considered a deviation from the original concentration of <10% acceptable.
Dilutional range
To assess the validity of diluting samples with dexamethasone concentrations greater than the upper calibrator, dexamethasone-free serum (n = 5) was spiked with dexamethasone to a final concentration of 32 nmol/L. The samples were then diluted with PBS 0.1% BSA to target concentrations of 2, 8, 16 and 24 nmol/L and assayed as described above. The measured concentration was then multiplied by the appropriate dilution factor and the deviation from the original concentration calculated. For all dilutions, we considered a deviation from the original concentration of <10% acceptable.
Carryover
Injection-to-injection carryover was assessed by sequentially injecting a pool of 40% methanol/water (vol/vol) spiked with 160 nmol/L dexamethasone followed by a pool of 40% methanol/water (vol/vol) (n = 3). Carryover <0.1% was considered acceptable.
Specificity
To investigate analytical specificity, solutions of structurally similar endogenous and exogenous compounds were spiked in to 40% methanol/water (vol/vol) to supra-physiological concentrations (i.e. 1000 nmol/L for endogenous compounds and 1 mg/L for exogenous compounds). The compounds tested included: 17-hydroxyprogesterone, 21-hydroxyprogesterone, 11-deoxycortisol, aldosterone, androstenedione, cortisol, corticosterone, cortisone, pregnenolone, testosterone, epitestosterone, dhea, dheas (spiked to 100 μmol/L), budesonide, prednisolone, betamethasone, beclomethasone, fludrocortisone, fludrocortisone acetate, fluocinolone acetonide and triamcinolone acetate. Any compound that produced a peak at the same retention time as any transition monitored would be considered significant and investigated further.
Stability and reproducibility
Stability of serum dexamethasone has previously been reported. 9 As such, we assessed the stability and reproducibility of the SLE extracts at 10°C (n = 35) and −20°C (n = 84). Two plates with anonymized samples were prepared as detailed above and analysed. Immediately postanalysis, the plates were retrieved, heat sealed and one stored at 10°C in the auto-sampler and the other at −20°C. After 96 h, the plates were vortex mixed for 1 min, and centrifuged at 880 × g for 5 min prior to re-analysis. The % difference between the paired results was calculated with a mean % difference of <10% considered acceptable.
Subjects and design
We recruited 95 postmenopausal females with the approval of the South Yorkshire Research Ethics Committee (REC Ref No. 11/H1310/2), who gave written informed consent for the study. Postmenopausal females were selected to minimize any binding protein associated matrix effects. The study was performed at the Clinical Research facility at Sheffield Teaching Hospitals NHS Foundation NHS Trust (study number: STH15803). No individuals were under investigation for CS or were known to have an AI. The mean (standard deviation [SD]) age of the cohort was 68.8 (6.3) years and the mean (SD) BMI was 27.1 (4.6) kg/m2. All participants undertook a 1 mg ONDST. The 1 mg tablet was administered at 23:00 and with a serum sample collected the following day between 08:00 and 09:00. 1 Samples were collected into 5-mL serum gel tubes. Serum was separated, aliquoted into 1.5 mL cryogenic vials and frozen at −20°C within 24 h. Samples were then distributed to Wythenshawe Hospital, Manchester University NHS Foundation Trust for analysis of cortisol and dexamethasone. Serum cortisol was measured using a previously published LC-MS/MS method, 15 and dexamethasone was measured as detailed above.
Data analysis
Analyse-It version 2.30 was used for data analysis.
Results
Following SLE extraction and LC-MS/MS analysis, dexamethasone and its deuterated internal standard eluted after 1.01 and 1.00 min, respectively (Figure 1). Both the m/z 393>237 and m/z 393 >147 transitions were found to be suitable transitions for quantitation. Although the m/z 393 > 147 transition was analytically more sensitive, the m/z 393 >237 transition was favoured due to superior results in the linearity experiments (data not shown).
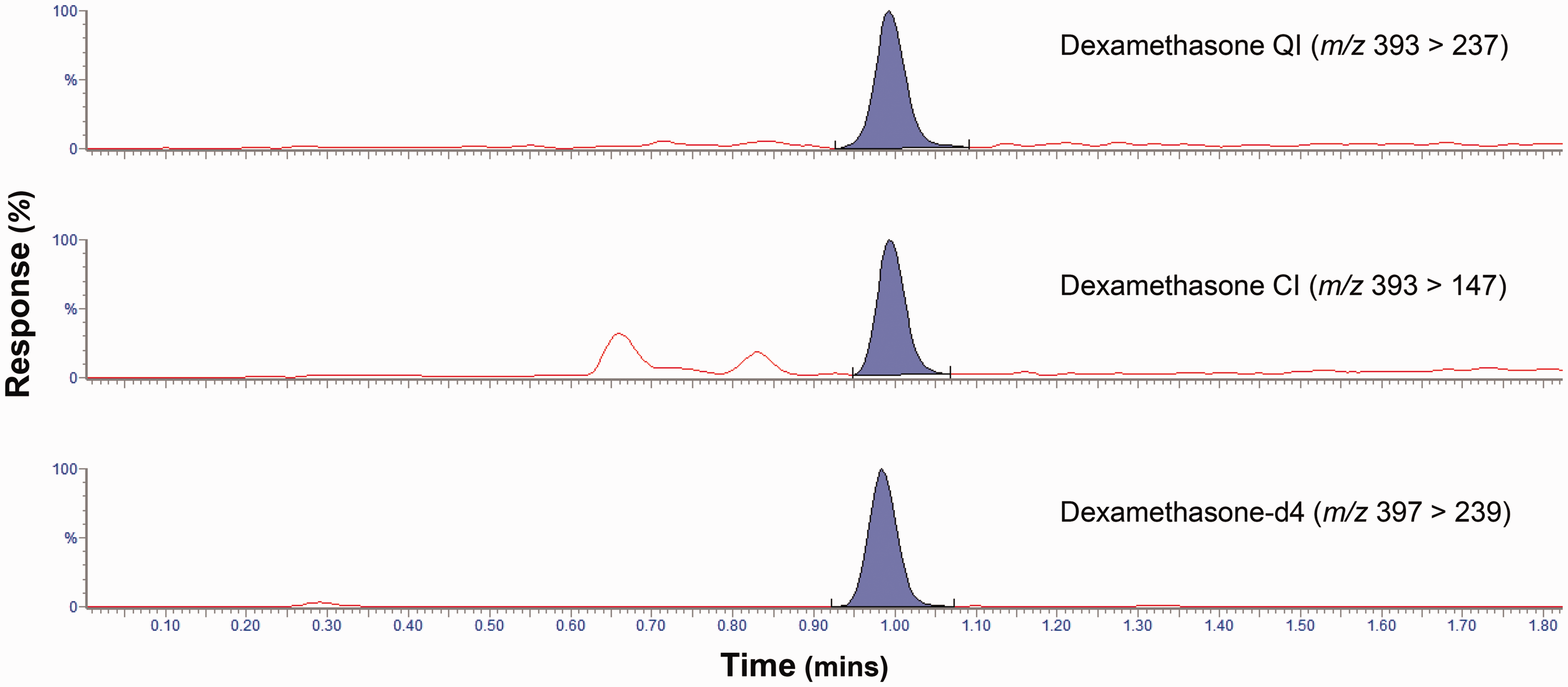
A typical chromatogram showing the response of the quantifying ion (QI), confirmatory ion (CI) and dexamethasone-d4 internal standard for a serum sample determined to have a dexamethasone concentration of 0.5 nmol/L.
Accuracy, as assessed from the deviation of observed dexamethasone concentrations from their target concentrations, was mean (range) 1.7% (−4.7 to 6.7) at 1.5 nmol/L, 2.8% (−2.2 to 9.3%) at 6.0 nmol/L and 1.6% (−2.3 to 5.7%) at 12.0 nmol/L. The method of standard additions confirmed PBS 0.1% BSA as a suitable matrix for calibrators and QCs with agreement between the two matrices <5%.
Multiple injections of the same QC materials yielded intra-assay imprecisions of 4.9%, 4.1% and 3.3% at concentrations of 1.5, 6.0 and 12.0 nmol/L, respectively. The inter-assay imprecision for the same QC materials was 3.2%, 3.4% and 2.2%, respectively.
The mean recovery ranged from 96 to 100%. Assessment of the mean matrix effect yielded results ranging from 106 to 109%; comparison of samples spiked pre-extract to postextract showed the mean extraction efficiency to be 94 to 98%. The mean overall process efficiency ranged from 101 to 105%.
Analysis of serum spiked with low concentrations of dexamethasone provided an LOQ of 0.25 nmol/L; at this concentration, a signal-to-noise of >5:1 was maintained, while the mean bias was found to be +5% and the imprecision was 7.3%. Sequential injections of 40% methanol/water (vol/vol) pools spiked with 160 nmol/L of dexamethasone followed by dexamethasone-free 40% methanol/water (vol/vol) found injection-to-injection carryover to be ≤0.04%.
Reanalysis of SLE extracts following storage at 10°C (n = 35) and −20°C (n = 84) for 96 h found the mean difference between results to be 6.5% (95% CI 5.3 to 7.7) and −5.5% (95% CI −6.2 to −4.9), respectively.
Standard curves were constructed by plotting dexamethasone concentrations on the x-axis and dexamethasone/dexamethasone-d4 peak height ratios on the y-axis. The curve was linear (R2≥0.99) over the calibration range of 0–16 nmol/L. Examination of the curve over 10 consecutive batches, produced a mean R2 value of 0.9996 (95% CI: 0.9993–0.9998) showing that linearity was reproducible between batches. Linearity on dilution experiments confirmed the relationship between analyte concentration, and response was linear over the desired assay range and that PBS 0.1% BSA was a suitable diluent for any samples with dexamethasone concentrations up to at least 32 nmol/L.
From the interference panel, peaks were observed in both the QI and CI channels for beclomethasone and betamethasone. The retention time for the beclomethasone peak was later than that of dexamethasone (1.09 vs. 1.01 m) and would therefore be readily identifiable in routine use. Conversely, the retention time of betamethasone was very close to that of dexamethasone (0.99 vs. 1.01 m). To investigate if this interference would be significant at pharmacological concentrations, we spiked betamethasone at a purity of ≥98% (Sigma, Poole, UK) into dexamethasone-free serum to a final concentration of 63 nmol/L (25 ng/mL). 16 Aliquots (n = 8) of the betamethasone spiked serum were then prepared and analysed as described above. We observed peaks in both the dexamethasone QI and CI channels at retention times of 0.99 m in each of the extracts which equated to a mean (SD) dexamethasone concentration of 4.75 (0.17) nmol/L. However, examination of the QI:CI ratio revealed betamethasone to have a mean ratio of 0.331 (95% CI: 0.328–0.334). Conversely, analysis of serum dexamethasone samples (n = 54) had a mean QI:CI ratio of 0.707 (95% CI: 0.703–0.712) which was similar to the mean QI:CI ratio of PBS 0.1% BSA calibrators and QC materials (n = 8) of 0.693 (95% CI: 0.662–0.725) (Table 1). Thus, the earlier retention time and use of a ±15% QI:CI ion ratio tolerance from the calibrator provides a robust means to identify betamethasone interference.
QI:CI ion ratios of dexamethasone in PBS 0.1% BSA and serum samples.

Note: The QI:CI for betamethasone spiked serum is also shown (samples were spiked to a final betamethasone concentration of 63 nmol/L [25 ng/mL]).
BSA: bovine serum albumin; PBS: phosphate-buffered saline; QI: quantifying ion; CI: confirmatory ion.
Analysis of 95 postmenopausal female samples revealed a total of 84/95 patients both suppressed cortisol to ≤50 nmol/L and had a dexamethasone concentration >3.3 nmol/L post-ONDST. A further three patients (3%) adequately suppressed cortisol to <50 nmol/L yet had a dexamethasone concentration >3.3 nmol/L. Of the 87 that achieved adequate suppression, the median (range) cortisol concentration was 25 nmol/L (13–47 nmol/L) and the median (range) dexamethasone concentration was 7.3 nmol/L (2.1–24.0 nmol/L).
A total of 8/95 patients failed to adequately suppress cortisol <50 nmol/L. Of these, three (3%) achieved a dexamethasone concentration >3.3 nmol/L (range 3.5 to 14.7) and a cortisol concentration close to the <50 nmol/L cut-off (range 51.8 to 53.5 nmol/L). Thus, five (5%) had inadequate suppression (cortisol >50 nmol/L) and dexamethasone ≤3.3 nmol/L (Figure 2). None of these five individuals were taking any medication that may influence the metabolism of dexamethasone. Of the five, four were found to have detectable concentrations of dexamethasone ranging from 1.36 to 2.84 nmol/L with cortisol concentrations ranging from 57.9 to 317.2 nmol/L. One individual was found to have an undetectable dexamethasone concentration (<0.25 nmol/L) and a corresponding cortisol concentration of 277.3 nmol/L.
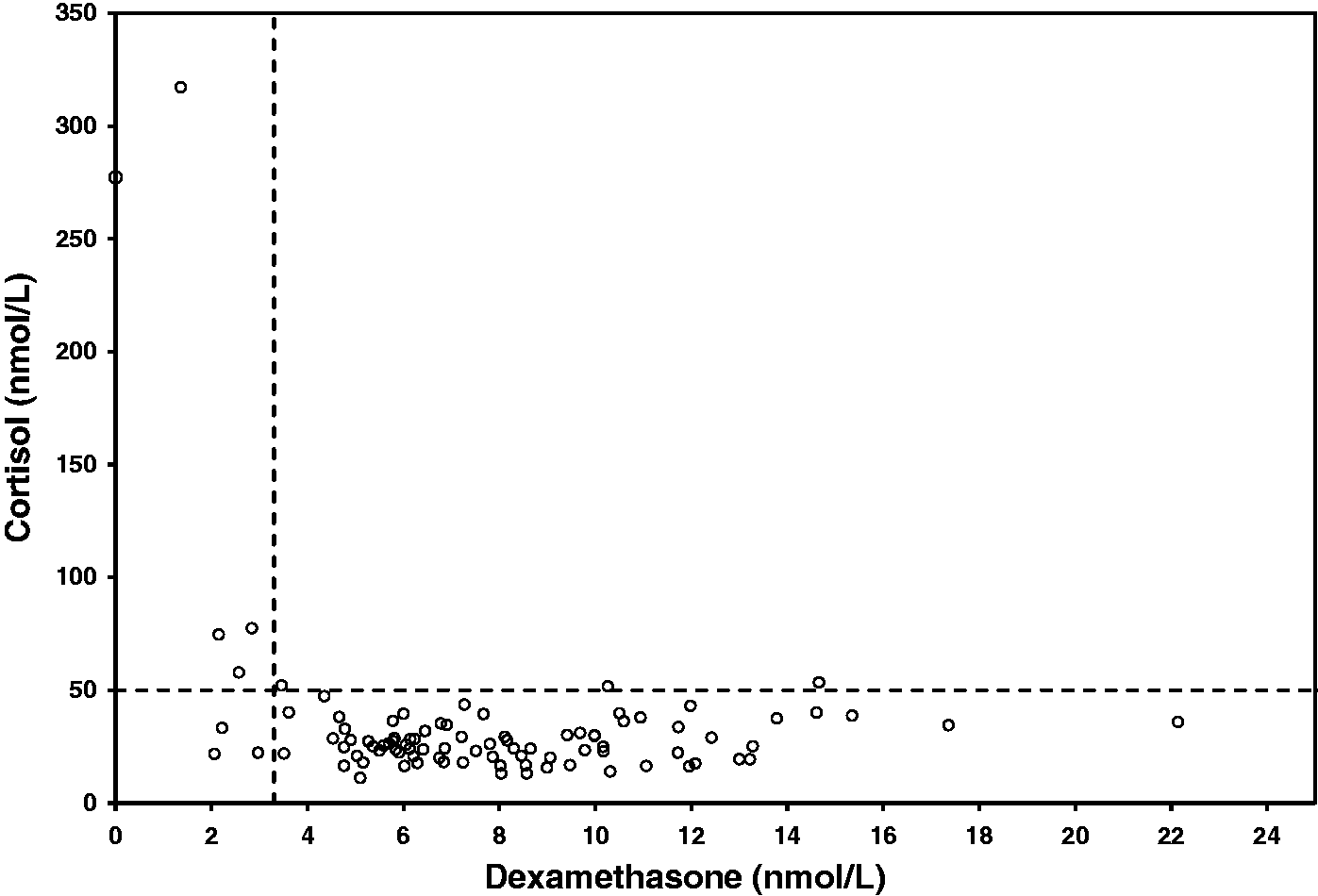
Dexamethasone and cortisol concentrations post 1 mg ONDST for the 95 participants. The dashed lines represent the cut-offs for results >3.3 nmol/L of dexamethasone and ≤50 nmol/L of cortisol.
Discussion
We have developed a rapid and robust LC-MS/MS method for serum dexamethasone quantitation. The assay benefits from using CRM for calibration thereby ensuring results are metrologically traceable to SI units through an unbroken chain of calibration.
In the absence of a reference measurement procedure or matrix-matched CRMs, the assay performance characteristics have been assessed using gravimetrically prepared materials spiked with various concentrations of dexamethasone. In all instances, our results have been acceptable when assessed against an industry standard.12,13
The use of SLE plates for sample purification provides a robust 96-well support that minimizes the retention of undesirable compounds such as salts, phospholipids and proteins. This helps to maximize analytical recovery and reduce matrix effects, thus allowing for an efficient and sensitive method capable of measuring dexamethasone down to concentrations of 0.25 nmol/L. SLE methods hold an advantage over SPE5–8 as they are considerably quicker, require fewer steps (thereby reducing inter-operator variation) and are generally less expensive. In addition, the SLE extracts are stable for up to 96 h when stored at either 10°C or −20°C allowing samples to be re-injected or temporarily stored should there be unforeseen instrument downtime.
Utilization of a short fused-core C8 column for chromatography allows both good separation and fast re-equilibration times. This has been integral to achieving an injection-to-injection run time of only 2.2 min. Although our method is unable to baseline separate dexamethasone from its stereoisomer betamethasone, we can reliably identify the presence of betamethasone through the use of ion ratios. Importantly, it is highly unlikely that patients being investigated for CS and autonomous cortisol secretion would be prescribed an exogenous glucocorticoid. Indeed, exclusion of iatrogenic CS secondary to exogenous glucocorticoid use is an essential consideration in the diagnostic work-up of CS prior to undertaking any additional investigations. 1
Endocrine Society guidelines suggest that measurement of serum dexamethasone and cortisol post-ONDST may help identify false-positive responses. 1 However, there remains a lack of normative data against which to substantiate this and, with different dexamethasone cut-offs of >5.63 and >3.3 nmol/L 4 proposed, there is a requirement to further verify this in order to provide the correct interpretation.
One concern surrounding the >5.6 nmol/L cut-off is that it was originally derived using an RIA method to quantitate both dexamethasone and cortisol. 3 Although once state of the art, RIA methods have subsequently been found to suffer from poor specificity. 17 Thus, the traceability of this cut-off and its application to more accurate mass-based quantitation may be compromised. Moreover, as this cut-off is based on defining adequate suppression post-ONDST using the classical cortisol cut-off of <138 nmol/L, 3 it should not by applied to post-ONDST cortisol concentrations ≤50 nmol/L.
Conversely, the >3.3 nmol/L dexamethasone cut-off has been determined using the recommended ≤50 nmol/L post-ONDST cortisol cut-off. 4 Accordingly, using our method to measure dexamethasone in 95 healthy postmenopausal females and applying the serum dexamethasone cut-off >3.3 nmol/L, we observed similar results to Ueland et al. 4 Whereas Ueland et al. identified 3% of subjects both failed to suppress serum cortisol ≤50 nmol/L and had a serum dexamethasone ≤3.3 nmol/L, we found 5% (5/95) of our population fulfilled this criterion. As none of the five were taking any medication known to induce CYP3A4, it is likely that these individuals may be fast metabolizers, poor absorbers or simply may not have complied with the protocol. None of the five went on to develop CS.
Our study supports the use of a post-ONDST dexamethasone cut-off of >3.3 nmol/L and may help identify false-positive tests. This method allows integration into the workflow of a busy, high-throughput laboratory and can help provide important clinical information before proceeding onto more expensive investigations. Should a patient fail the ONDST and be found to have a dexamethasone concentration ≤3.3 nmol/L, we recommend either repeating the test after checking for confounding factors (compliance or re-checking for CYP3A4 inducing medication/agents) or performing a 48-h low-dose dexamethasone suppression test. Depending on the clinical situation, alternatives would include performing another recommended screening test such as a 24-h urine free cortisol collection or midnight salivary cortisol collection.1,2
In summary, we have developed a rapid LC-MS/MS method for serum dexamethasone quantitation. The performance characteristics of the assay are acceptable when judged against an industry standard.12,13 The assay is ISO 15189 accredited and can be used to help differentiate false-positives from true-positives in the investigation of CS and AI or for pharmacokinetic studies.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship or publication of this article.
Ethical approval
The South Yorkshire Research Ethics Committee (REC Ref No. 11/H1310/2) approved the study.
Guarantor
JMH.
Contributorship
BGK, LJO, MD and JN-P conceived the study. JMH, LJO and BGK developed the method. MD and JN-P gained ethical approval and conducted the study. JMH wrote the first draft of the manuscript. All authors reviewed and edited the manuscript and approved the final version.