
Abstract
Variegate porphyria is an autosomal dominant acute hepatic porphyria characterized by photosensitivity and acute neurovisceral attacks. Hepatocellular carcinoma has been described as a potential complication of variegate porphyria in case reports. We report a case of a 48-year-old woman who was diagnosed with hepatocellular carcinoma following a brief history of right upper quadrant pain which was preceded by a few months of blistering lesions in sun-exposed areas. She was biochemically diagnosed with variegate porphyria, and mutational analysis confirmed the presence of a heterozygous mutation in the protoporphyrinogen oxidase gene. Despite two hepatic resections, she developed pulmonary metastases. She responded remarkably well to Sorafenib and remains in remission 16 months after treatment. A review of the literature revealed that hepatocellular carcinoma in variegate porphyria has been described in at least eight cases. Retrospective and prospective cohort studies have suggested a plausible association between hepatocellular carcinoma and acute hepatic porphyrias. Hepatic porphyrias should be considered in the differential diagnoses of hepatocellular carcinoma of uncertain aetiology. Patients with known hepatic porphyrias may benefit from periodic monitoring for this complication.
Introduction
The porphyrias are a group of heterogeneous disorders resulting from acquired or genetically determined enzyme deficiencies of the haem biosynthetic pathway. Variegate porphyria (VP; OMIM 176200), an autosomal dominantly inherited porphyria, is consequent to a partial deficiency of protoporphyrinogen oxidase (PPOX), the penultimate enzyme in haem biosynthesis. 1 In common with hereditary coproporphyria (HCP), it may present with acute neurovisceral attacks and cutaneous involvement with approximately 60% of patients with VP presenting with skin lesions, 20% with acute neurovisceral attacks alone and 20% with both these manifestations. VP is about one-third as prevalent as acute intermittent porphyria (AIP) in most European countries. 2 In Europe, the VP population is genetically heterogeneous with low penetrance, and only a minority of patients experience recognizable symptoms potentially leaving many at risk of long-term sequelae. Hepatocellular carcinoma (HCC) has been reported as a complication of VP. 3 We discuss the case history of a patient with HCC manifesting within a few months of her first cutaneous lesions of undiagnosed VP.
Case history
A 48-year-old Caucasian woman presented to her general practitioner (GP) with dyspepsia and right upper quadrant abdominal pain. A few months prior to this, she had noticed blistering lesions on the dorsa of both hands. She was otherwise fit and well with no active medical problems. There were no risk factors for liver disease, i.e. minimal alcohol intake, no recent foreign travel, no previous blood transfusions and no significant family history. Clinical examination revealed right upper quadrant fullness but was unremarkable otherwise. An ultrasound scan of the liver revealed a 14-cm predominantly echogenic heterogeneous mass in the liver with limited blood flow. The spleen, pancreas, gallbladder, kidneys and abdominal aorta were normal. A follow on computerized tomography (CT) scan delineated a well-defined mass in the right hepatic lobe measuring 11 × 14 cm in maximal axial dimension. The rather chaotic internal enhancement characteristics necessitated further imaging by magnetic resonance imaging (MRI), and this suggested the presence of HCC. 4 The serum alpha fetoprotein (AFP) was >300 kU/L (reference interval: 3–8 kU/L, Immulite). Human Haemochromatosis Protein (HFE) gene testing found that she was a C282Y heterozygote with normal ferritin and transferrin saturation. Serology testing for infective and autoimmune liver disease was negative. She was referred to the hepatobiliary surgeons. In view of the cutaneous lesions, she was also initially referred to dermatology and subsequently to the specialist porphyria clinic.
Investigation and management
Porphobilinogen and porphyrin methods
Total urine porphyrins were measured spectrofluorimetrically after oxidation of any porphyrinogen. Total faecal porphyrins were measured spectrophotometrically on the Hitachi U-2001 after acid extraction of porphyrins from faeces. Porphyrin isomers were separated and measured directly from acidified urine or extracted faeces by reversed-phase gradient elution chromatography on the Shimadzu high-performance liquid chromatography (HPLC) system. Plasma porphyrins were qualitatively scanned on the Hitachi F-4500 fluorescence spectrophotometer. Free porphyrins were extracted from plasma and analysed fluorimetrically using an excitation scan. The plasma porphyrin concentration was calculated using a ‘GWBASIC’ computer programme which corrects for background interference. The calibration material was sourced from independent suppliers, and the excitation and emission wavelength accuracy was checked weekly to ensure optimum performance using the sharp spectral line of the xenon lamp.
Porphobilinogen and porphyrin results
The initial porphyrin screen included urine, faeces and plasma samples which showed:
Urine porphyrin/creatinine ratio: 146.1 nmol/mmol (<28) Faecal porphyrins: 459 nmol/g dry weight (<130) Faecal coproporphyrin III/I ratio: 4.0 (<2.0) Plasma poprhyrins: 231 nmol/L (<11.2 nmol/L with an excitation maximum at 401 nm and an emission maximum at 623 nm)
While these results were clearly abnormal and highly suggestive of porphyria, the low plasma porphyrin scan emission maximum and presence of numerous interfering peaks within both the urine and faecal porphyrin HPLC fractionation scans raised sufficient concern for the laboratory to request repeat samples and recommend further evaluation in the porphyria clinic. Five months after initial presentation, she was reviewed in the porphyria clinic and was noted to have crusted blisters and milia suggestive of cutaneous porphyria on the dorsa of both hands. Further samples of plasma, urine and faeces showed:
Urine porphobilinogen (PBG): 0.42 µmol/mmol (<0.9) Urine porphyrin/creatinine ratio: 23.4 nmol/mmol (<28) Faecal porphyrins: 222 nmol/g dry weight (<130) Faecal coproporphyrin III/I ratio: 2.8 (<2.0) Plasma porphyrins: 13 nmol/L L (<11.2 nmol/L with an excitation maximum at 405 nm and an emission maximum at 626 nm)
The plasma porphyrin scan and inverse faecal coproporphyin III/I ratio supported the earlier findings and were consistent with a phenotype of VP.
Porphyria genetic investigation
DNA sequencing analysis of the exons 1–13 including the flanking intronic regions of the PPOX gene confirmed the presence of a heterozygous sequence variant c.1292_1293delAG. This previously described pathogenic mutation causes a frame shift leading to a premature STOP codon with consequent deficient enzyme activity. 5 Genetic counselling and presymptomatic testing was recommended for the family members of the index case. Patient education was provided for triggers of an acute attack, information on safe drugs to use in porphyria and skin protection.
Further management
Three months after first presentation, she had an uneventful right hemi-hepatectomy. Histology confirmed a solitary HCC with vascular invasion but no features of cirrhosis or steatosis in adjacent liver. Special stains were negative for iron, copper-associated protein and alpha-1-antitrypsin deficiency. Postoperative liver imaging and biochemical surveillance were satisfactory for the initial nine months. Albumin and bilirubin monitoring demonstrated minimal variation, while alkaline phosphatase rose only moderately in relation to hepatic surgery. Renal function was relatively well preserved with creatinine ranging between 55 and 92 µmol/L (estimated Glomerular Filtration Rate (eGFR) 63–>90 ml/min/1.73 m2). Ten months postoperatively, serum AFP concentrations were noted to have risen (see Figure 1). Serial imaging by CT, MRI and positron emission tomography (PET) did not identify any evidence of local or distant recurrence for a further three months.
Serial monitoring of serum bilirubin, ALT, ALP and AFP. The figure shows biochemical response of various biomarkers to treatment.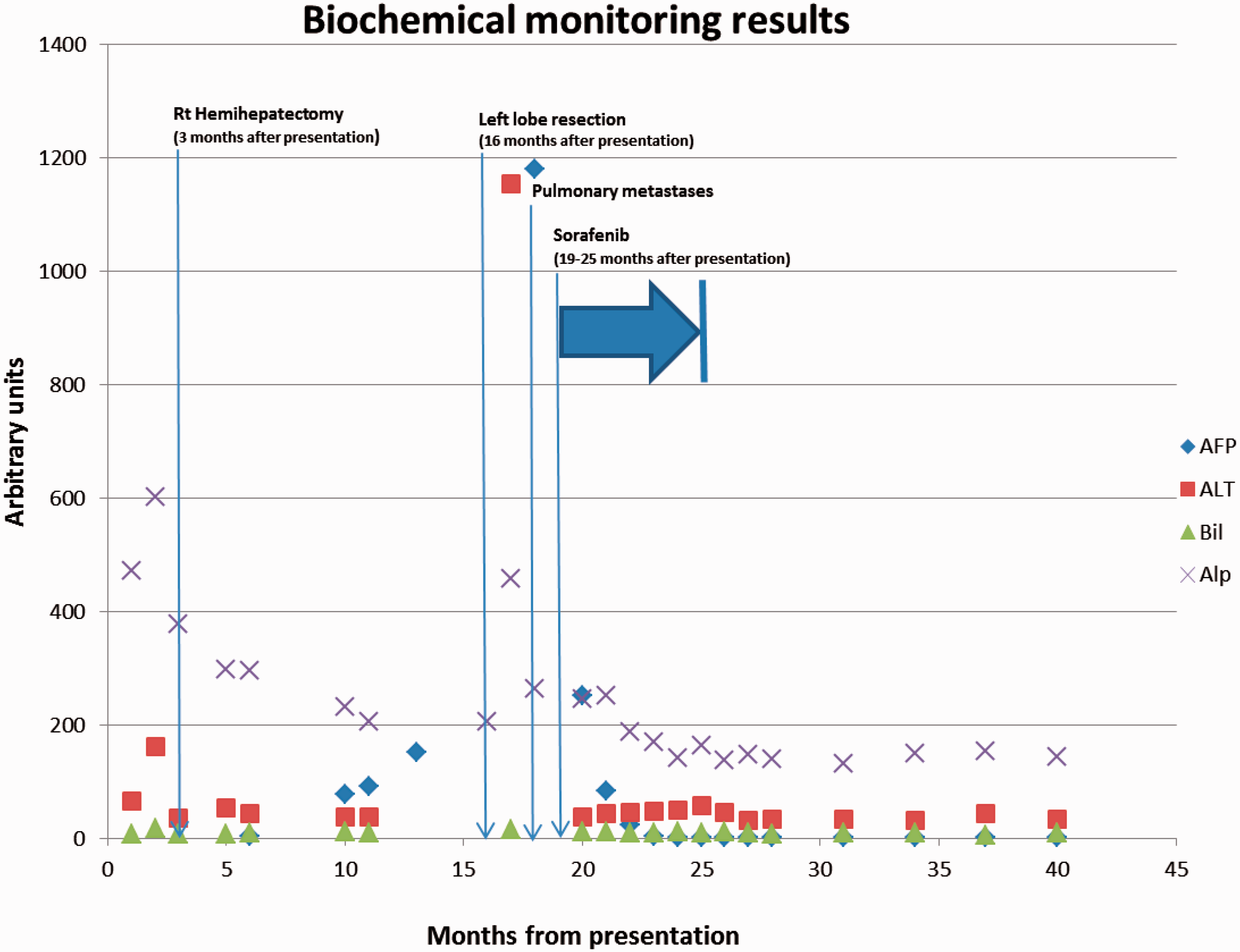
Thirteen months after her initial hemi-hepatectomy, a PET scan identified a new fluorodeoxy glucose (FDG) avid 5.1 cm mass in the remnant liver consistent with a solitary local recurrence of HCC. A left lobe resection was performed, and the histology confirmed a moderately poorly differentiated recurrent HCC with no surrounding cirrhosis. The vascular invasion was extensive in the recurrence. Although the original right hemi-hepatectomy had a very close resection margin, the recurrence was in the tip of the left lobe, and therefore more consistent with a separate tumour than a margin recurrence from the first resection. Immunohistochemistry for Cytokeratin 19 (CK19) which is associated with aggressive tumour behaviour was negative in both original and recurrent tumours. One month after her second surgery, she became dyspnoeic. Her AFP and alanine aminotransferase (ALT) were found to have risen markedly. CT images confirmed multiple pulmonary nodules representing disseminated lung metastases.
She was commenced on a six-month cycle of Sorafenib by the medical oncology team. Within three months of commencing this treatment, her AFP normalized, and CT imaging showed marked resolution of the pulmonary metastases with only two small nodules still visible. At completion of the six-month course of treatment, CT imaging demonstrated complete resolution of her lung metastases, while her AFP remained within reference limits. At the point of writing this case report, she remained in clinical, radiological and biochemical remission 16 months after completing her treatment with Sorafenib.
Discussion
Summary of the reported cases of HCC in VP.
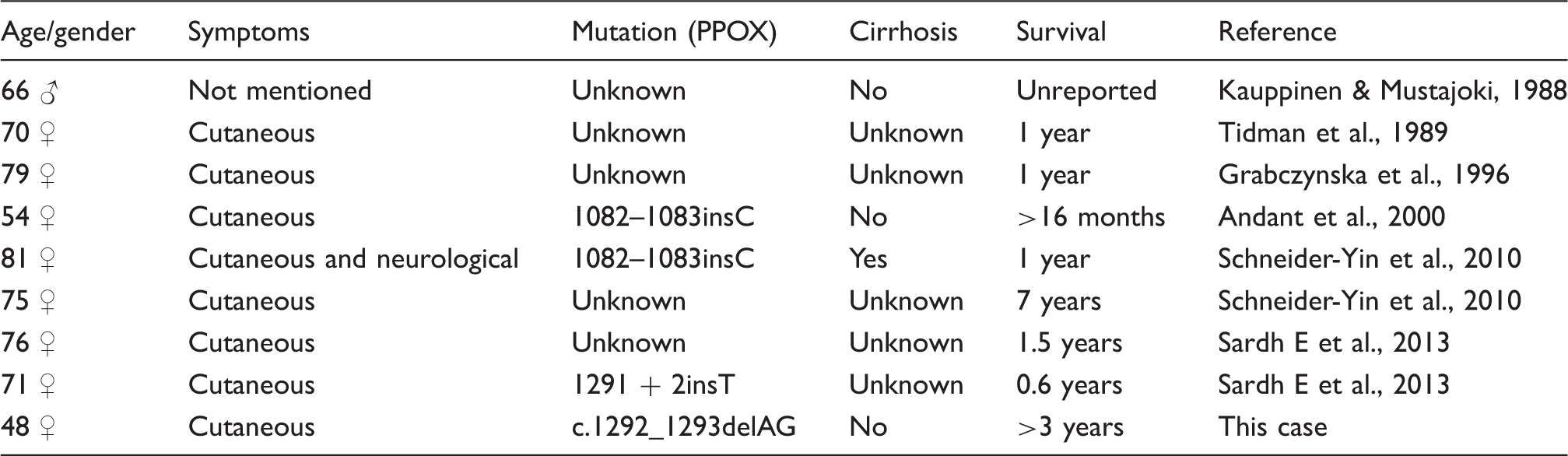
This case report adds to the previously reported cases in supporting a plausible link between HCC and VP particularly when traditional risk factors for HCC are absent. Given the rarity of VP, case reports represent an important way of highlighting these observations as more rigorous evaluation is likely to be hampered by low enrolment and/or lack of outcome events. A notable observation is that eight of the nine known cases had VP diagnosed at around the same time as the HCC. This may indicate that HCC in VP differs aetiopathogenetically from AIP. It may even be argued that the HCC induces a first presentation of VP in previously asymptomatic gene carriers. The reasons underlying this co-presentation remain to be discovered, but there are important clinical implications for patients newly diagnosed with either condition. The limitations of case reports must be considered. The observations reported are not controlled for chance, bias or confounders, and therefore they are at best used for generating hypotheses. In particular, causal inference, generalization and over interpretation should be avoided. 16 More robust data are emanating from multicentre cohorts such as the prospective study from the European Porhyria Network (EPNET) which supported a link between HCC and VP. 2
Our case highlights the difficulties of diagnosing porphyria especially in non-specialist centres. The initial screening results though clearly abnormal and suggestive of porphyria were rendered uncertain by the presence of interfering peaks in the urine/faeces HPLC fractionation scans and a low plasma porphyrin scan emission maximum. These ultimately led to several months delay in the diagnosis of VP. Further investigations were not undertaken to identify the source of the interfering peaks, but it is suspected that the likely source was medication. Several drugs, most notably quinolones, vitamin-enriched yeast tablets and riboflavin from multivitamin supplements have been reported to produce interference probably due to their fluorescence emission spectra overlapping those of various porphyrins. 17 Our patient was not on antibiotics at the time of porphyrin testing; however, the possibility of exposure to over-the-counter vitamin supplements cannot be discounted. While this did not affect the long-term outlook, there was potential for missing the diagnosis entirely if repeat testing was not undertaken. It is worth noting that porphyria diagnosis should include urine, faeces and plasma samples. Additionally, repeat testing is essential if the quality of results is in question.
The prognosis of HCC is reliant on early diagnosis and effective therapeutic intervention. Early-stage HCC yields five-year survival rates of up to 60–70% because it is amenable to potentially curative treatments such as surgical therapies (resection and liver transplantation) and locoregional procedures (radiofrequency ablation). 18 Advanced stages have a dismal prognosis as there are limited effective treatment options. 19 The SHARP study demonstrated that Sorafenib (an oral multikinase inhibitor) increased median survival and time to radiologic progression by three months in patients with advanced HCC and relatively well-preserved liver function. Overall median survival for treated patients was 10.7 months. 20 Our patient is unusual in that she has already survived >3 years since her diagnosis, 16 months of which are after Sorafenib therapy. This must be balanced against the observation that a previous HCC/VP patient survived for seven years after diagnosis; and the rare but reported incidence of spontaneous regression of HCC which is estimated at 0.4%. 21
Conclusion
Our patient together with the others reported in the literature highlights the importance of considering VP as a risk factor for HCC. Debate continues about the cost effectiveness of screening for HCC in acute hepatic porphyria patients with the argument in favour of screening in AIP.22,23 For VP, screening may be less beneficial as the two conditions often present together. Furthermore, the exercise may not be feasible or cost-effective because there are no programmes to follow-up asymptomatic gene carriers in the UK. Clinicians should, however, be aware of the possibility of both conditions being present when either VP or HCC is newly diagnosed, especially if traditional hepatic risk factors are absent. The relative younger age of our patient compared to those in the literature (48 years versus >50 years at diagnosis of HCC) raises the possibility that investigating for HCC in newly diagnosed VP should be considered even in patients below 50 years.
Footnotes
Acknowledgements
We would like to thank Sharon Whatley, Cardiff Porphyria Service for genetic analysis.
Declaration of conflicting interests
None declared.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethical approval
Written consent to publication obtained from patient.
Guarantor
JHB.
Contributorship
AL wrote the first draft. All authors were involved in the management of the patient and reviewed the manuscript.