
Abstract
Niemann–Pick disease, type B (NPD-B; OMIM 607616) is an inborn error of metabolism where reduced concentrations of the enzyme acid sphingomyelinase (ASM; EC 3.1.4.12) lead to multisystem disease though with survival into adulthood. The natural history of NPD-B is one of progressive hypersplenism and gradual deterioration of pulmonary function. We describe a 46-year-old South African man of French Huguenot descent who presented to a lipid disorders clinic with mixed hyperlipidaemia. Clinical examination and imaging findings revealed the presence of massive hepatosplenomegaly, interstitial lung disease and subclinical atherosclerosis; there were no neurological or cognitive abnormalities. Laboratory testing showed thrombocytopaenia, increased liver transaminases and mild hyperbilirubinaemia. Lysosomal enzyme analysis showed markedly reduced ASM activity, suggestive of NPD. DNA sequence analysis of the SMPD1 gene revealed that he was a compound heterozygote for the previously reported c.1829_1831delGCC (ΔR608) mutation and a novel missense mutation c.1378A > C (p.T460P). In conclusion, we describe the clinical findings of a case of NPD-B with mixed hyperlipidaemia, compound heterozygous for the SMPD1 ΔR608 mutation and a novel mutation, T460P.
Introduction
Niemann–Pick disease type B (NPD-B; OMIM 607616) is a rare, autosomal recessive, lysosomal storage disease due to deficient activity of acid sphingomyelinase (ASM; EC 3.1.4.12), which results in accumulation of sphingomyelin within cells and tissues of affected individuals.1,2 It is pan-ethnic and results from allelic mutations in the SMPD1 gene. A non-neuronopathic disease, it is characterised by hepatosplenomegaly, an atherogenic lipid profile, and progressive pulmonary involvement. 3 Liver dysfunction, cardiac disease, retinal stigmata and growth restriction may also occur. However, the disease manifestations are variable, with most patients surviving into adulthood. 4
We describe the clinical findings of a case of NPD-B with mixed hyperlipidaemia, compound heterozygous for the SMPD1 ΔR608 mutation and a novel mutation, T460P.
Clinical case
A 46 year-old South African man of French Huguenot descent was referred to a lipid disorders clinic with mixed hyperlipidaemia. The only significant past medical history was the lipid disorder and relatively limited exercise tolerance since childhood, with no deterioration over the past few years.
On physical examination, he looked of good health with a body mass index of 23 kg/m2 and BP 120/70 mmHg. There were no peripheral stigmata of lipid disorders. Auscultation of his heart and lungs revealed an accentuated second heart sound and crackles in the mid-zone and bases. There was massive hepatosplenomegaly, but no clinical signs of a coagulopathy. There were no neurological or cognitive abnormalities and fundoscopic examination was normal.
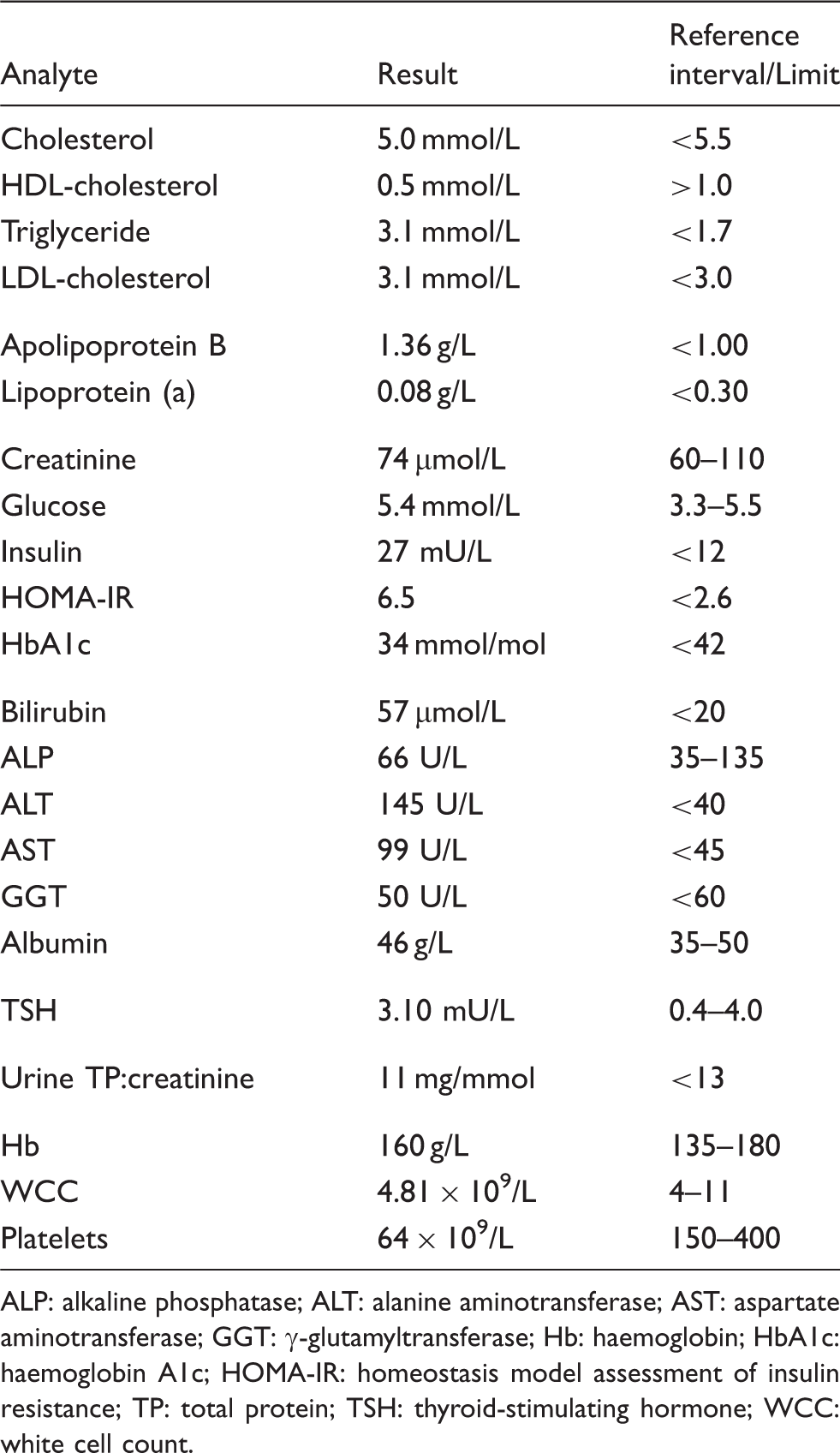
Biochemical testing revealed normal glucose, kidney and thyroid function. Laboratory testing confirmed the mixed hyperlipidaemia and showed thrombocytopaenia, increased liver aminotransferases and mild hyperbilirubinaemia, with normal hepatic synthetic capacity (Table 1). A computed tomography (CT) abdomen confirmed the hepatosplenomegaly, but also revealed extensive bilateral adrenal calcification as well as vascular calcification of the aorta, common iliac arteries and splenic artery, CT chest showed interstitial lung disease and CT coronary angiography revealed the presence of subclinical atherosclerosis with a coronary calcium score of 179, which was at the 97th percentile (Figure 1). A baseline serum cortisol was 270 nmol/L and 30 min Synacthen-stimulated cortisol was 460 nmol/L (normal response >430, Abbott Architect), excluding the possibility of adrenal insufficiency secondary to the adrenal calcification. Baseline electrocardiogram and stress echocardiography were normal. Pulmonary function tests showed a relative preservation of flow rate (FEV1 2.58 L; 75% of predicted) and lung volume (VA 4.6 L; 76% of predicted), with markedly decreased diffusion capacity for carbon monoxide (DLCO 9.9 mL/min/mmHg; 36% of predicted). Lysosomal enzyme analyses showed a markedly reduced ASM activity of 4 nmol/min/mg protein (20–120), consistent with the diagnosis of NPD, with normal activity of all the other enzymes analysed as part of the neurolipidosis screen.
Diagnostic imaging: (a) CT abdomen showing gross hepatosplenomegaly with a mass lesion within the lower pole of the spleen and a small focal lesion superiorly. There were no focal liver lesions, (b) CT abdomen showing extensive bilateral adrenal calcification as well as vascular calcification of the aorta, common iliac arteries and splenic artery, (c) CT chest showing diffuse ground-glass opacification with basal predilection and ‘crazy paving’ and (d) CT coronary angiography does not reveal any definite coronary stenosis; the coronary calcium score was 179 (97th percentile). Biochemical and haematological results. ALP: alkaline phosphatase; ALT: alanine aminotransferase; AST: aspartate aminotransferase; GGT: γ-glutamyltransferase; Hb: haemoglobin; HbA1c: haemoglobin A1c; HOMA-IR: homeostasis model assessment of insulin resistance; TP: total protein; TSH: thyroid-stimulating hormone; WCC: white cell count.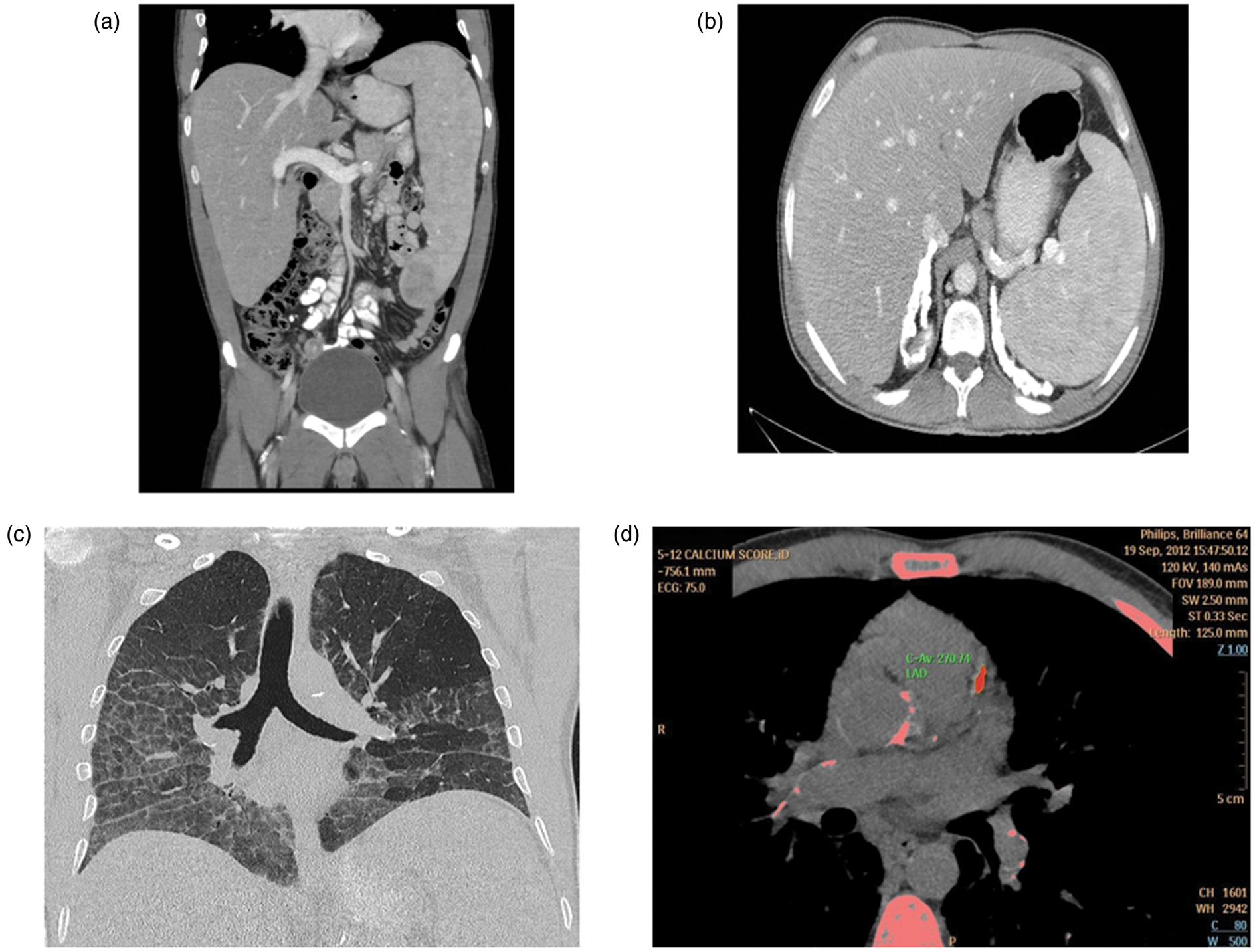
DNA sequence analysis of the SMPD1 gene revealed that he was a compound heterozygote for the previously reported c.1829_1831delGCC (Δ R608) mutation and a novel missense mutation c.1378A > C (p.T460P). We have been unable to undertake mutation analysis on the patient’s parents.
Discussion
The possibility of a lysosomal storage disorder should be considered in a patient with hepatosplenomegaly after excluding primary liver diseases, malignancies and haematological disorders. The most important lysosomal storage disorders in adults include: Gaucher’s disease (β-glucocerebrosidase deficiency), cholesterol ester storage disease (acid lipase deficiency) and NPD (ASM deficiency). NPD is a heterogeneous disorder with a wide clinical spectrum which correlates with residual ASM enzyme activity. Most affected NPD-B patients have thrombocytopaenia secondary to hypersplenism, which increases the risk of bleeding episodes.1,4 Other clinical manifestations include short stature with delayed skeletal maturation, interstitial lung disease, typically mild liver dysfunction, ocular and lipid abnormalities comprising low HDL-cholesterol and hypertriglyceridaemia. Over time there is progressive hypersplenism and deterioration of pulmonary function. In contrast to NPD-A, most patients with NPD-B have no neurologic abnormalities.
NPD-B is caused by mutations in the SMPD1 gene located on chromosome 11p15.1–p15.4. The gene contains 6 exons, encoding a 629 amino acid protein product. It has been identified that certain SMPD1 mutations follow a fairly mild, adult-onset form of NPD-B, whereas other mutations predict an early-onset, more severe form. This case report describes a mildly affected NPD-B patient, who was found to be compound heterozygous for ΔR608 and a novel missense mutation T460P. The ΔR608 mutation is a 3 bp deletion in exon 6 that results in the removal of an arginine residue from residue 608 on the ASM polypeptide. 5 While this mutation is prevalent in Ashkenazi Jews and North Africans, a large study in over 200 NPD-B patients worldwide showed that the ΔR608 mutation occurred on ∼12% of NPD-B alleles. 6 It has been suggested that the presence of the ΔR608 mutation is neuroprotective, 6 as patients with NPD-B have little or no central nervous system involvement, often surviving into adolescence or adulthood.
The novel missense mutation T460P affects an amino acid that is completely conserved throughout the kingdom Animalia. Algorithms Mutation Taster (http://www.mutationtaster.org/) and PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/index.shtml) predict T460P to be pathogenic. Amino acid 460 lies within the metallophosphoesterase catalytic domain (residues 257–461). 7 Together, this suggests that T460P is likely to be pathogenic. However, as the T460P mutation was found in a compound heterozygote with the neuroprotective ΔR608 mutation, the phenotypic severity of T460P is not clear.
Our case demonstrates many of the classical features of NPD-B with the clinical findings of hepatosplenomegaly, thrombocytopaenia, liver enzyme abnormalities, respiratory complications, and an atherogenic lipid profile – low HDL-cholesterol and high triglyceride and apolipoprotein (apo) B. 8 There was no evidence of a familial dyslipidaemia or premature cardiovascular disease. In the absence of severe hypertriglyceridaemia, secondary causes of marked hypocholesterolaemia, such as androgen use, malignancy, and primary monogenic disorders, namely apoA-I mutations, Tangier disease, and lecithin-cholesterol acyltransferase deficiency should be excluded if clinically indicated. 9 The defective ASM in NPD-B results in sphingomyelin enrichment of HDL which impairs HDL maturation and contributes to HDL deficiency.10,11 Although unknown, the mechanism for the dyslipidaemia in NPD-B may relate to over-secretion of very low-density lipoprotein owing to increased hepatic lipid availability and/or delayed clearance of apoB-containing lipoproteins from the circulation.
In conclusion, we describe the clinical findings of a case of NPD-B with mixed hyperlipidaemia, compound heterozygous for the SMPD1 ΔR608 mutation and a novel mutation, T460P. The mechanism for the atherogenic dyslipidaemia with NPD-B remains to be determined. Of note, a Phase 1 clinical trial is evaluating the safety and tolerability of recombinant human ASM as an enzyme replacement therapy in adults with NPD-B (NCT01722526).
Footnotes
Acknowledgements
JRB is supported by a Practitioner Fellowship from the Royal Perth Hospital Medical Research Foundation.
Declaration of conflicting interests
None declared.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Ethical approval
Not required. Written informed consent from the patient was obtained.
Guarantor
JRB.
Contributorship
YG wrote the first draft of the manuscript. All authors edited the manuscript and approved the final version of the manuscript.