
Abstract
Adenoviruses are extensively studied in terms of their use as gene therapy vectors and pathogenesis. These vectors have been targeted on both transcriptional and transductional levels to achieve cell-specific gene delivery. Current detection strategies, including reporter gene expression, viral component detection, and vector labeling with fluorophores, have been applied to analyze adenoviral vectors; however, these methods are inadequate for assessing transductional targeting. As an alternative to conventional vector detection techniques, we developed a specific genetic labeling system whereby an adenoviral vector incorporates a fusion between capsid protein IX and EGFP. DNA packaging and thermostability were marginally hampered by the modification while DNA replication, cytopathic effect, and CAR-dependent binding were not affected. The fluorescent label was associated with the virus capsid and conferred a fluorescent property useful in detecting adenoviral particles in flow cytometry, tracking, and tissue sections. We believe our genetic adenovirus labeling system has important implications for vector development, detecting adenovirus vectors in targeting schemes, and studying adenovirus biology. In addition, this technique has potential utility for dynamic monitoring of adenovirus replication and spread.
Introduction
Gene therapy offers promise for treating a variety of diseases. Common to all gene-based therapeutic approaches is the use of vectors to deliver a gene of interest to specific cells. An improved understanding of both viral and non-viral vector biology in recent years has afforded the possibility to achieve both enhanced uptake of vectors into resistant cells and targeted gene delivery to specific cells while avoiding non-target tissues [1]. Such restricted gene delivery can be attained by two means: transcriptional targeting through the use of tissue specific promoters [2,3] and transductional targeting through genetically modified or conjugate-based vectors [4]. Vector design based on these premises requires sensitive tools to validate the function of targeting.
Reporter gene expression is an excellent method to evaluate transcriptional targeting in which the promoter of interest could be coupled with a reporter in a chosen delivery vehicle. Typically for adenoviral vectors, reporter genes such as GFP (green fluorescent protein) [5], luciferase [6], somatostatin receptor type 2 (SSTR-2) [7], sodium iodide symporter [8], and herpes simplex virus thymidine kinase (HSV-TK) [9,10] have been applied for this purpose. Most of these techniques are not only operative in vitro but also show utility for whole-body detection in mice [11]. Reporter gene expression has also been employed to evaluate the function of targeting schemes based on the second strategy of vector transduction. Although generally useful for this purpose in vitro, reporter gene expression does not truly represent the physical biodistribution of adenoviral vectors following systemic administration. in vivo, the bulk of vectors sequestered by the innate and acquired immune system as well as nontarget cells may result in degradation rather than productive transduction to induce reporter transgene expression [12–15]. Understandably, transgene expression relies not only on successful cellular entry but also on effective gene delivery to the nucleus. Therefore, reporter gene expression cannot be reliably utilized to analyze differential biodistribution as a result of targeting.
To more accurately detect vectors in situ, techniques have been used to physically probe for viral components, including DNA and protein. Methods such as immunofluorescence or in situ hybridization, however, demand extensive preparation and strong accessible signals to achieve visualization. Additionally, polymerase chain reaction amplification, while highly sensitive, cannot provide spatial information at the cellular level. An alternative means to physically localize vectors is through direct labeling with detectable chemical substrates. One way to accomplish vector labeling is by using fluorophores such as fluorescein isothiocyanate (FITC) [16], Texas Red [17], and carbocyanine (Cy3) [18]. This technique has proven to be very powerful in the study of intracellular trafficking of adenoviral vectors and has also been used to discern the localization of vectors after systemic injection [12,15,19]. Nevertheless, the general utility of this system for the assessment of vector targeting remains questionable due to several reasons. Labeling adenoviruses with synthetic dyes involves additional steps of conjugation and dialysis or centrifugation to remove unbound fluorophore [16,18], which may affect the yield and quality of the virus [15,20,21]. In the case of FITC labeling, more than 40% of the dye was incorporated into capsid proteins other than hexon including the fiber and penton base [16]. Nonspecific labeling also occurs with Texas Red, whereby a fraction of the dye is also conjugated to fiber and penton base [17]. Unfortunately for Cy3, the sites of fluorophore incorporation have not been evaluated. Ultimately, these modifications of the adenovirus capsid may alter its charge and binding character, possibly affecting in vivo biodistribution and clearance, therefore confounding targeting strategies [22].
To address these issues, we hypothesized that a system whereby adenoviruses are physically linked with a fluorescent structural protein label during replication would provide progeny virions with a unique fluorescent property useful for particle detection. Recently, we successfully incorporated heterologous peptide sequences onto the carboxy-terminus of the external capsid cement protein IX (pIX) of human adenovirus type 5 [23]. We used a similar strategy to link enhanced green fluorescent protein (EGFP) to the virus capsid by fusing pIX with EGFP, thereby achieving specific genetic labeling of the capsid at this particular locale. The results show that this fluorescent structural label was incorporated into the adenovirus with minimal effect on viral function and that the viral particles have a fluorescent property exploitable for detection in adenoviral vector targeting studies both in vitro and in situ.
Materials and Methods
Cell Culture
Human embryonic kidney 293 (American Type Culture Collection [ATCC], Manassas, VA), human embryonic retinoblast 911 [24], human lung adenocarcinoma A549 (ATCC), and Chinese hamster ovary (CHO) cells were maintained according to the suppliers' protocols. Cells were incubated at 37°C and 5% CO2 under humidified conditions.
Recombinant Adenovirus Construction
All viruses were constructed by homologous recombination in E. coli [25]. Control virus Ad-CMV-EGFP expresses EGFP (BD Biosciences. Clontech, Palo Alto, CA) from the deleted E1 region and contains native IX. Ad-IX-EGFP was constructed from a modified pShuttle vector called pShlpIXNhe (AdEasy system, Qbiogene, Carlsbad, CA), which has the IX gene with a C-terminus FLAG tag linker followed by a unique NheI restriction site [23]. The EGFP HincII-SacI fragment from pABS.4-CMV-tetR-EGFP (with a stop codon) was blunted with large Klenow fragment (New England Biolabs, Beverly, MA) and inserted in frame into pShlpIXNhe which was linearized with NheI and also blunted. A correct clone (called pShuttle-IX-EGFP), based on restriction digest and expression of pIX-EGFP, was linearized with PmeI and homologously recombined with pAdEasy-1 (AdEasy system, Qbiogene) in electrocompetent BJ5183 to generate an adenoviral backbone containing IX-EGFP in place of wild-type IX. The shuttle plasmid (pShuttle-wt-IX-EGFP) for production of Ad-wt-IX-EGFP (wild-type adenovirus with IX-EGFP fusion gene) was constructed by inserting the SgrAI-MfeI fragment from pXC1 (Microbix, Toronto, Canada) containing the E1 region as wellas a 5′ portion of IX into pShuttle-IX-EGFP, also digested with SgrAI and MfeI. pShuttle-wt-IX-EGFP was homologously recombined with pTG3602 to generate a wild-type adenoviral genome with IX-EGFP in place of wild-type IX. The final backbones were linearized with PacI and then transiently transfected into 911 cells (Superfect, QIAGEN, Valencia, CA) for virus propagation. Both Ad-CMV-EGFP and Ad-IX-EGFP are E1- and E3-deleted while Ad-wt-IX-EGFP contains both E1 and E3 (Fig. 1A).
Virus Propagation and Purification
Viruses were propagated in 911 cells and 293 cells which express very low levels of wild-type pIX that are insufficient for complementing pIX deficiency (J. Vellinga, personal communication and data not shown) [26–28]. Viruses were purified by double cesium chloride (CsCl) ultracentrifugation and dialyzed against phosphate-buffered saline with Mg2+, Ca2+, and 10% glycerol. Final aliquots of virus were analyzed for viral particle titer (absorbance at 260nm) and transducing unit titer (t.u.). Based on a previously described protocol [12], t.u. was determined by infecting 911 cells in 96-well plates with 1/10 serial dilutions of the virus and counting the number of green cells 2 days postinfection (n = 6). All viruses were stored at −80°C until use.

pIX-EGFP intracellular localization. (A) Control Ad-CMV-EGFP construct with native IX and Ad-IX-EGFP construct with IX-EGFP in place of native IX. (B) EGFP cellular localization from Ad-CMV-EGFP and pIX-EGFP almost exclusive nuclear localization from Ad-IX-EGFP.
DNA Packaging Analysis
DNA packaging was analyzed by using a previously reported protocol [29]. Briefly, 911 cells (5 × 104) were infected with Ad-CMV-EGFP and Ad-IX-EGFP at 10 t.u./cell in 12-well plates. On Days 1,2, 3, and 4 postinfection, the cells and medium were collected, processed for DNA (QIAamp DNA Blood Mini Kit, QIAGEN), and analyzed with Taqman quantitative real-time PCR using E4 specific primers (LightCycler™ System, Roche Applied Science, Indianapolis, IN). One half of the cell pellet was analyzed for total intracellular viral genome copy number and the other half for intracellular encapsidated viral genome copy number. The supernatant was analyzed for released encapsidated viral genome copy number.
Cytopathic Effect Assay
Ten thousand 293 cells or 911 cells were infected with Ad-CMV-EGFP and Ad-IX-EGFP (10, 1, and 0.1 t.u./cell) in 100 μL of 1% and 2% DME medium without Phenol red, respectively (six replicates for each condition plus six noninfected wells as controls). Cytopathic effect was measured by an MTS assay (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, Promega, Madison, WI) on Days 0, 2, 4, 6, 8, and 10 postinfection. Results were calculated as percent of noninfected cells with blank values (medium only) subtracted beforehand.
Thermostability Assay
Thermostability was analyzed using a modified version of a previously reported protocol [23]. Samples of Ad-CMV-EGFP and Ad-IX-EGFP (106 t.u. in 100 μL PBS) were incubated for various time periods at 45°C (0, 5,10, 20, and 40 min). Transducing unit infectious titers were then determined for the samples using the abovementioned protocol.
Taqman Real-Time Quantitative PCR Binding Assay
All steps were carried out with 1% BSA/PBS buffer. CHO and A549 cells (2 × 105 in 100 μL) were incubated with 100 μL buffer alone or buffer containing 2.5 μg/mL recombinant fiber knob at 4°C with vigorous shaking. After one hour, Ad-CMV-EGFP or Ad-IX-EGFP was directly added (5000 moi based on genome copy number determined by Taqman quantitative PCR using E4 primers). Following shaking for another hour with the viruses at 4°C, the cells were washed three times, collected for total DNA preparation (QIAamp DNA Blood Mini Kit, QIAGEN), and then quantitated using Taqman quantitative real-time PCR with E4 primers as mentioned above.
Characterization of Virus Gradient Fractions
For fractionation studies, Ad-CMV-EGFP and Ad-IX-EGFP were propagated each in ten 150-mm dishes. Both viruses were purified by CsCl ultracentrifugation where the top and bottom bands were retained after two centrifugation steps yielding one gradient from the 10 dishes. After the second spin, fractions (~100 μL) were collected dropwise through a perforation at the bottom of the tube. Each fraction (50 μL) was diluted with 150 μL PBS in microcuvettes and measured for relative fluorescence with a Versafluor fluorometer (BioRad, Hercules, CA) using 490/10 nm excitation and 510/10 nm emission filters (Chroma Technology, Brattleboro, VT). To determine viral DNA content, a sample of each fraction (10 μL) was diluted in 90 μL 0.5% SDS/PBS and incubated at room temperature for 10 min to release the viral genomes. Absorbance at 260 nm was then measured for each sample (MBA 2000, Perkin Elmer, Shelton, CT).
Western Blot
Fractionated samples (4 μL) were ethanol precipitated, pelleted, and resuspended with 20 μL RIPA buffer. Samples (5 μL) were resolved with SDS-PAGE and then transferred to a polyvinylidene difluoride (PVDF) membrane (BioRad, Hercules, CA). Blotting was performed with a primary monoclonal GFP antibody (1:1000 dilution, BD Biosciences Clontech) followed by a secondary HRP-linked anti-mouse antibody (1:5000 dilution, Amersham Pharmacia, Piscataway, NJ) or a primary polyclonal hexon antibody (1:1000 dilution, Chemicon International, Temecula, CA) followed by a secondary HRP-linked anti-goat antibody (1:5000 dilution, Dako, Carpinteria, CA). Bands were detected with a chemiluminescent ECL kit (Amersham Pharmacia Piscataway, NJ).
Fluorescence Microscopy
Epifluorescence microscopy was performed with an inverted IX-70 microscope (Olympus, Melville, NY) equipped with a Magnifire digital CCD camera (Optronics, Goleta, CA). Glass slides and coverslips were used to mount samples (Fisher Scientific, Pittsburgh, PA). Images were acquired with a × 100 objective using oil immersion (2sec exposure) and processed with Adobe Photoshop 7.0 (San Jose, CA). Hoescht staining (Molecular Probes, Eugene, OR) was used to visualize nuclear DNA.
Flow Cytometry Virus Binding Assay
Incubation and blocking methods similar to the Taqman quantitative real-time PCR binding assay were used. The sCAR-EGF conjugate (recognizing the epidermal growth factor receptor) [30] was used to demonstrate CAR-independent binding to A549 cells. Prior to incubation, Ad-IX-EGFP was conjugated with sCAR-EGF for one hour at room temperature (12 pmol per 5 × 109 vp). Conjugated or nonconjugated Ad-IX-EGFP (5000 vp/cell) was incubated with 5 × 105 CHO [CAR(–) and EGFR(–)] or A549 [CAR(+) and EGFR(+)] cells in 200 μL 1% BSA/PBS at 4°C. A549 cells blocked beforehand with recombinant fiber knob were washed prior to addition of sCAR-EGF-conjugated Ad-IX-EGFP. After incubation with the viruses, the cells were washed three times and then analyzed by flow cytometry with EGFP detection. Gates were set according to data obtained for cells not incubated with virus.
Tracking of Ad-IX-EGFP Infection
A549 cells (1 × 105) were plated on 30mm glass-bottom cell culture dishes (World Precision Instruments, Sarasota, FL) the day prior to infection with growth medium lacking phenol red (1% FCS/DME medium). The cells were incubated for one hour at 37°C with or without recombinant adenovirus fiber knob (1 μg/mL) in 500 μL of growth medium. After Hoescht staining for 5 min and three washes with the above medium, Ad-IX-EGFP (10000 vp/cell) in 300 μL growth medium supplemented with 25 mM Hepes was added to the cells for 30 min at 4°C. Following three washes to remove unbound virus, images of early infection were then acquired using the above fluorescence microscopy technique. Images of late infection were acquired after incubating the dish for another hour at 37°C.
In situ Detection of Ad-wt-IX-EGFP
All methods were approved by and performed according to the guidelines of the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham. C57/BL6 mice (Charles River Laboratories, Wilmington, MA) were anesthetized with 10 mg each of ketamine and xylazine, and an abdominal incision was made to expose the inferior vena cava into which Ad-wt-IX-EGFP (1011 vp) was injected. The mice were sacrificed 20 min (C57/BL6) following virus injection. Frozen sections (5 μm thick) of the liver were fixed onto glass slides and stained with the macrophage F4/80 antibody (1:50 dilution, 4 ng/μL final concentration, Caltag, Burlingame, CA) followed by goat anti-rat secondary antibody (1:100 dilution, 6 ng/μL final concentration, Caltag). The section was also stained with Hoechst for nuclear DNA. Glass coverslips were mounted on the slides with 90% glycerol/PBS and then imaged as described above. For the targeting study, B6CBAF1C57/hCAR transgenic mice (human coxsackie adenovirus receptor transgenic mice, kind gift from Dr. S. Pettersson, Center for Genomics Research, Stockholm, Sweden) [31] and B6CBAF1/J wild-type control mice (Jackson Labs, Bar Harbor, ME) were used. Mice were sacrificed 10 min after Ad-wt-IX-EGFP injection into the inferior vena cava with the same amount of virus as above. Lung and liver frozen sections (5 μm thick) were stained with Hoechst and visualized using the above procedure.
Results
Expression of pIX-EGFP Fluorescent Label
An E1-deleted shuttle plasmid was constructed containing a IX-EGFP carboxy-terminal fusion gene in place of wild-type IX. Following transient transfection of this plasmid into 911 cells, pIX-EGFP was expressed and accumulated in intranuclear inclusions as previously reported [32] (data not shown). This shuttle plasmid was used to construct E1- and E3-deleted Ad-IX-EGFP (Fig. 1A), which was successfully rescued. In an adenoviral context, pIX-EGFP was expressed and also accumulated in the nucleus. In contrast, Ad-CMV-EGFP infected cells showed diffuse localization of EGFP (Fig. 1B).
DNA Packaging Efficiency of Ad-IX-EGFP
Protein IX has been shown to play various roles in adenovirus infection, including capsid stabilization, transcriptional activity, and nuclear reorganization [32,33]. Although dispensible in packaging [26], adenovirus pIX is important in packaging full-length genomes and stabilizing the capsid structure [27,34]. To characterize the effect of fusing EGFP to pIX on DNA packaging and viral yield, viral DNA from various fractions was quantitated using Taqman quantitative PCR on Days 1, 2, 3, and 4 following infection of 911 cells at 10 t.u./cell. These fractions included intracellular total, intracellular encapsidated, and supernatant encapsidated viral DNA. The results indicate that total viral DNA replication was the same for both Ad-IX-EGFP and control Ad-CMV-EGFP. However, both packaged viral genomes in the cell and packaged viral genomes released into the supernatant were approximately half a log factor lower for Ad-IX-EGFP compared to the control (Fig. 2A), indicating lower progeny yield.
Cytopathic Effect of Ad-IX-EGFP
To quantitatively evaluate viral cytoxicity, infection of 911 and 293 cells with Ad-IX-EGFP and control virus at 10, 1, and 0.1 fcu/cell multiplicities of infection (moi) was monitored over 10 days. On Days 0, 2, 4, 6, 8, and 10, the cytopathic effect of the virus was quantitated using a nonradioactive cell proliferation MTS assay. In both 293 cells (data not shown) and 911 cells, Ad-IX-EGFP cytopathic effect was the same as that of Ad-CMV-EGFP (Fig. 2B). These findings suggest that pIX-EGFP did not affect the cytopathic capacity and lateralization of the virus.
Thermostability of Ad-IX-EGFP
Modification of pIX may destabilize the capsid structure. This possibility was assessed by comparing the thermostability of Ad-IX-EGFP to that of control Ad-CMV-EGFP. Virus samples were incubated at 45°C for various time periods and then quantitated in terms of infectious titer. Up to 5 min of incubation, the infectious titer of Ad-IX-EGFP remained the same as that of control. After 10 min of incubation, the infectious titer of Ad-IX-EGFP was about 50% lower than that of Ad-CMV-EGFP. By 20 and 40 min of incubation, the titer of Ad-IX-EGFP was not significantly different from that of Ad-CMV-EGFP (Fig. 3A). These results indicate that the EGFP addition to pIX slightly affected the thermostability of Ad-IX-EGFP following intermediate exposure of the virus to high temperature.
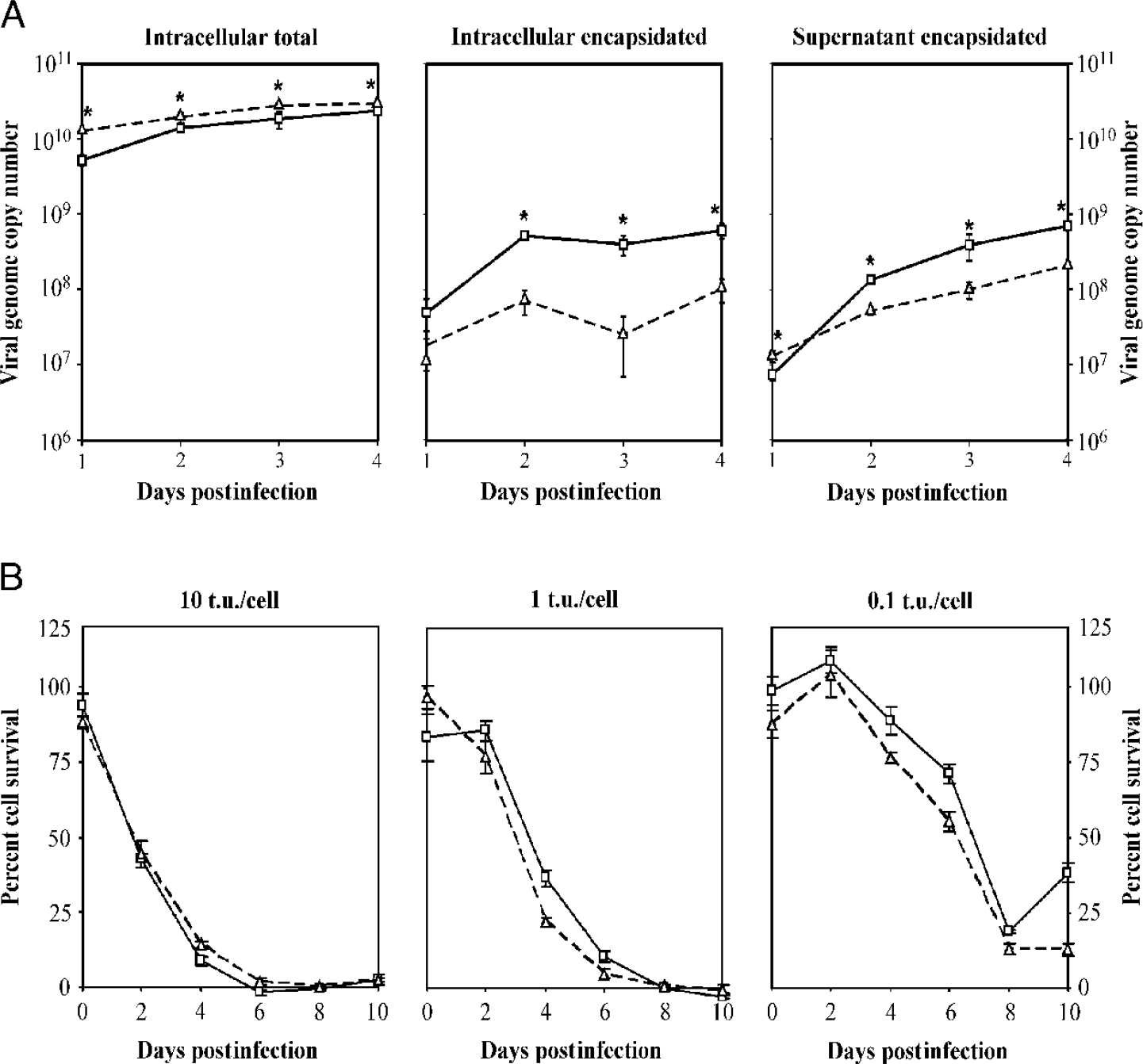
Characterization of Ad-IX-EGFP. (A) DNA packaging efficiency of Ad-IX-EGFP. Postinfection viral genome copy numbers from various pools (intracellular total, intracellular encapsidated, and supernatant encapsidated) for Ad-CMV-EGFP (—□—) and Ad-IX-EGFP (—△—) (n = 3). *Viral genome copy numbers were statistically different (single factor ANOVA, p < .05) between the two viruses. (B) Cytopathic effect and spread of Ad-IX-EGFP. Time-course cytopathic effect of Ad-CMV-EGFP (—□—) and Ad-IX-EGFP (—△—) in 911 cells (very low pIX expression) at various multiplicities of infection. Cytotoxicity measured by a nonradioactive proliferation assay and expressed as percent survival (percentage of living cells relative to noninfected cells, n = 6). T.u. = transducing unit infectious titer.

Characterization of Ad-IX-EGFP. (A) Thermostability of Ad-IX-EGFP. Both viruses were incubated at 45°Cfor various time periods and then quantitated in terms of transducin unit infectious titer (n = 3). White bar = Ad-CMV-EGFP and gray bar = Ad-IX-EGFP. *p < .05 between the two bars at 10 min (single-factor ANOVA). (B) CAR-dependent binding of Ad-IX-EGFP. After binding to CHO CAR(–) cells and A549 CAR(+) cells, bound viral genome copy numbers were quantitated by Taqman real-time quantitative PCR (n = 3). Several groups were initially blocked with recombinant fiber knob. White bar = Ad-CMV-EGFP and gray bar = Ad-IX-EGFP. *p < .05 between the two bars (single-factor ANOVA). T.u. = transducing unit infectious titer.

Incorporation of pIX-EGFP into Ad-IX-EGFP particles. (A) Fluorescence (○) and DNA content (▪) of Ad-CMV-EGFP and Ad-IX-EGFP CsCl gradient fractions following double ultracentrifugation. Fractions were also analyzed using anti-GFP and anti-hexon Western blotting after SDS-PAGE. (B) Visualization of Ad-IX-EGFP particles. Dilutions (1:100) of representative top and bottom band fractions for Ad-CMV-EGFP and Ad-IX-EGFP were visualized with epifluorescence microscopy using a × 100 oil immersion objective.
CAR-Dependent Binding of Ad-IX-EGFP
The coxsackie adenovirus receptor binding ability of Ad-IX-EGFP was determined by allowing the virus to bind to cells and quantitating the bound virus with Taqman quantitative real-time PCR. Ad-IX-EGFP binding to CAR(+) A549 cells was much higher than that to CAR(–) CHO cells. Recombinant fiber-knob block mitigated the CAR-dependent binding of Ad-IX-EGFP to A549 cells. Blocking had no effect on the binding of Ad-IX-EGFP to CHO cells. This CAR-dependent binding trend was very similar to that of Ad-CMV-EGFP (Fig. 3B). Overall, Ad-IX-GFP appeared to show greater binding compared to Ad-CMV-EGFP. These results indicate that the capsid fusion protein label did not interfere with the ability of Ad-IX-EGFP to recognize its primary receptor CAR.
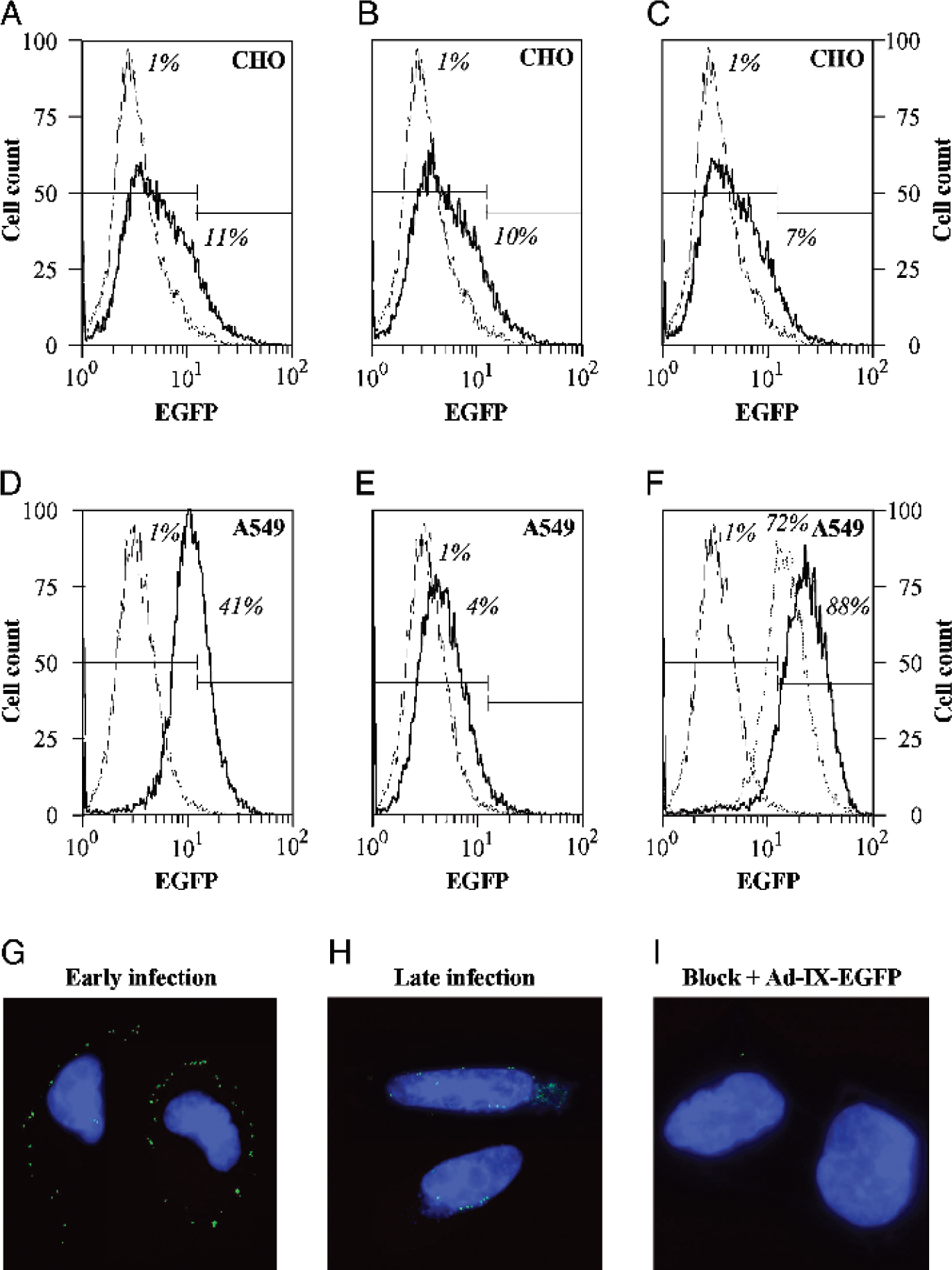
Application of Ad-IX-EGFP in binding and tracking assays. Ad-IX-EGFP particles bound to CHO [CAR(–) and EGFR(–)] and A549 [CAR(+) and EGFR(+)] cells were detected by flow cytometry. Blocking was performed with recombinant fiber knob while CAR-independent binding was tested with an sCAR-EGF conjugate. The thin lines are CHO or A549 cells only without Ad-IX-EGFP while the dark lines represent the following cases: (A) CHO + Ad-IX-EGFP; (B) CHO + knob + Ad-IX-EGFP; (C) CHO + sCAR-EGF + Ad-IX-EGFP; (D) A549 + Ad-IX-EGFP; (E) A549 + knob + Ad-IX-EGFP; (F) A549 + sCAR-EGF + Ad-IX-EGFP. In F, the dashed line corresponds to A549 + knob + wash + sCAR-EGF + Ad-IX-EGFP. Percentages given represent the fraction of cells positive for bound virus. Ad-IX-EGFP infection was tracked in live A549 cells. (G) A549 cells imaged at early infection (~20 min at 37°C after addition of virus). (H) A549 cells imaged at late infection (~1.5 hr at 37°C after addition of virus). (I) A549 blocked with recombinant fiber knob before infection. Blue = Hoechst stain for nuclear DNA.
Incorporation of pIX-EGFP into Ad-IX-EGFP Particles
Propagated Ad-CMV-EGFP and Ad-IX-EGFP were purified by double CsCl ultracentrifugation where the top and bottom bands were preserved. The resulting gradients of virus were fractionated and analyzed for fluorescence by fluorometry and DNA content by absorbance at 260 nm. Fluorescent peaks were observed for the top and bottom band fractions of Ad-IX-EGFP which were significantly greater than those of control Ad-CMV-Luc (data not shown) and Ad-CMV-EGFP (more than 2 log factors). For all viruses, a major DNA content peak was present in the bottom band where mature, infectious virions normally migrate (Fig. 4A). Infectious, mature Ad-IX-EGFP particles were present in the bottom band as indicated by an infectivity peak in a quick CPE assay (data not shown). The same gradient fractions of Ad-IX-EGFP and Ad-CMV-EGFP were also analyzed with Western blots using GFP and hexon antibodies. The blots show accumulations of pIX-EGFP and hexon in both the top and bottom band regions for Ad-IX-EGFP, confirming the colocalization of pIX-EGFP with the capsid and hence the virions. While hexon was also detected both in the bottom and top bands for Ad-CMV-EGFP, EGFP was only present above the top band where cellular proteins migrate (Fig. 4A).
Visualization of Ad-IX-EGFP Particles
Fluorescence microscopy of Ad-IX-EGFP bottom and top band fractions revealed abundant punctate green particles. The fluorescence of bottom band mature particles was uniform, whereas the fluorescence of top band empty particles had variable intensities. No fluorescent particles were detected for control Ad-CMV-Luc (data not shown) and Ad-CMV-EGFP (Fig. 4B).

In situ detection of Ad-wt-IX-EGFP in mouse liver. Ad-wt-IX-EGFP (1011 viral particles) was injected into the inferior vena cava of C57/BL6 mice followed by sacrifice at 20 min postinjection. Liver frozen sections (5 μm thick) were stained for Kupffer cells and nuclear DNA. (A) Ad-wt-IX-EGFP particles (green). (B) Stain for Kupffer cells with macrophage antibody (red). (C) Merged image of A and B. (D) Zoomed in image of box demarcated in A, B, and C. Arrows indicate pIX-EGFP signal resembling individual particles.
Detection of Ad-IX-EGFP Binding in a Flow Cytometry Assay
In addition to fluorescence microscopy, the possibility of Ad-IX-EGFP detection by flow cytometry was also evaluated. The viruses were allowed to bind to cells which were either blocked or not blocked beforehand with recombinant adenovirus fiber knob. Washed cells were then analyzed by flow cytometry, detecting for EGFP. Similar to the data obtained in the Taqman real-time PCR binding assay, Ad-IX-EGFP showed minimal binding (which was not affected by recombinant fiber-knob blocking [11% and 10% of cells positive for bound virus, respectively, Fig. 5A and B]) to CAR(–) CHO cells. Ad-IX-EGFP CAR-dependent binding to CAR(+) A549 cells was clearly detected which was abrogated by recombinant fiber-knob blocking (41% and 4%, respectively, Fig. 5D and E). Retargeted, CAR-independent binding was assayed using Ad-IX-EGFP conjugated with an sCAR-EGF fusion protein which mediates binding of the virus to the epidermal growth factor receptor (EGFR) [30]. Enhanced virus binding was detected for EGFR(+) A549 cells when sCAR-EGF was used (88% with sCAR-EGF compared to 41% without sCAR-EGF, Figure 5F, dark line, and Figure 5D, respectively) but not for EGFR(–) CHO cells (7%, Fig. 5C). The retargeted, CAR-independent binding to A549 cells was more apparent when the cells were blocked with knob initially and then washed before the addition of sCAR-EGF conjugated Ad-IX-EGFP (72% with block and sCAR-EGF, Fig. 5F, dashed line, compared to 4% with block alone, Fig. 5E).
Tracking of Ad-IX-EGFP Infection
Ad-IX-EGFP infection was monitored to demonstrate another utility of the optical property of this virus. Ad-IX-EGFP was allowed to bind to A549 cells at 4°C for 30 min and then imaged to show localization of the virus on the cell membrane early in infection (Fig. 5G). After allowing the infection process to continue for another hour at 37°C, the cells were imaged again to show perinuclear localization of the virus late in infection (Fig. 5H). These observations indicate that the pIX-EGFP label did not affect the internalization and trafficking of the virus. A549 cells blocked initially with recombinant fiber-knob showed minimal infection by Ad-IX-EGFP (Fig. 5I), again confirming the preservation of CAR-dependent binding of the virus.

In situ detection of Ad-wt-IX-EGFP in a targeting system. Ad-wt-IX-EGFP was injected intravenously into a wild-type control mouse and a transgenic hCAR mouse of the same strain which ubiquitously expresses truncated hCAR. Ten minutes after injection, the mice were sacrificed, and the lungs were sectioned and stained for nuclear DNA. (A) Wild type mouse lung. (B) hCAR mouse lung. (C) Zoomed in image of box demarcated in A. (D) Zoomed in image of box demarcated in B. For all panels, blue = Hoechst stain for nuclear DNA. For C and D, dashed lines delineate edges of lung tissue within which are areas of brown autofluoresence; orange arrows pointing diagonally right indicate lung yellowish-green autofluorescence from connective tissue; and white arrows pointing diagonally left specify bright green Ad-wt-IX-EGFP signal, some resembling single particles.
Detection of Ad-wt-IX-EGFP in Tissue
The application of our labeled adenovirus would be greatly expanded if it could be used in vector biodistribution and pathogenesis studies. To demonstrate the detectability of Ad-wt-IX-EGFP in tissues, the virus was injected into the inferior vena cava of a C57/BL6 mouse (1 × 1011 vp). Twenty minutes after virus injection, the mouse was sacrificed, and frozen sections (5 μm thick) of the liver were stained for Kupffer cells with a macrophage antibody as well as for nuclear DNA with Hoechst. Conglomerates of Ad-wt-IX-EGFP particles were easily detected in Kupffer cells (labeled red) along the sinusoids (Fig. 6A, B, and C) while pIX-EGFP signal resembling individual particles was visible in the parenchyma, indicative of hepatocyte infection (Fig. 6D). Similar to previously reported data [35,36], most of the virus localized to the Kupffer cells which are the principal cells that clear adenovirus in vivo while much fewer particles were found in the parenchyma of the liver.
Detection of Ad-wt-IX-EGFP in a Targeting System
To further illustrate the utility of our genetically labeled adenovirus, the fluorescent virus was applied in a targeting model to observe biodistribution differences under targeted versus nontargeted circumstances. Targeting was analyzed from the standpoint of an established, stringent animal model where we knew a priori the accessibility of the receptor in the target organ. For this purpose, a transgenic mouse was chosen which expresses a truncated human coxsackie-adenovirus receptor ubiquitously. As previously reported, this hCAR mouse represents a targeting context where enhanced localization of adenovirus to the lungs is expected relative to a wild-type control mouse [31]. Ad-wt-IX-EGFP was injected into the inferior vena cava of the hCAR mouse and its wild-type control (1 × 1011 vp). Ten minutes after virus injection, the mice were sacrificed, and frozen sections (5 μm thick) of the liver and lung were stained for nuclear DNA with Hoechst. In agreement with previously reported data [31], very few green adenoviral particles localized to the wild-type mouse lung (Fig. 7A and C, white arrows), whereas an abundance of labeled viruses distributed to the hCAR mouse lung (Fig. 7B and D, white arrows). These fluorescent particles were distinguishable from the yellowish-green autofluorescence due to connective tissue in the lung (orange arrows). Many labeled virions were also detected in both the wild-type and hCAR mouse livers (data not shown), resembling the results shown above (Fig. 6A-D). These data demonstrate the feasibility of detecting our genetically labeled adenoviruses in situ in a targeting biodistribution study.
Discussion
An appeal of adenovirus for gene therapy is the ability to target this vector through transcriptional and transductional means to achieve cell-specific and enhanced gene delivery. To evaluate the function of these targeting maneuvers, detection methods based on reporter gene expression, viral component localization, and vector labeling have been employed. Despite the various applications of these techniques, their broad utility for evaluating vector targeting has been challenged by a number of limitations. The ideal instrument for assessing distribution of viral vectors is one that gives a signal directly associated with the viral particles themselves. As an alternative to the conventional method of nonspecifically conjugating adenovirus with synthetic fluorescent dyes, we sought to develop a genetic vector labeling system that provides specific labeling of the adenovirus capsid with minimal compromise on vector function. The strategy we pursued involved the fusion of a fluorescent protein with the capsid protein pIX. The reasons for selecting this locale include its function as a minor capsid protein that is not absolutely required for replication, its externally exposed carboxy terminus to which proteins may be fused without affecting the interior of the capsid, and its distant location in the hexon group of nine which would not affect the penton base as well as the fiber-knob.
We have shown herein that our novel genetic labeling strategy for adenovirus marginally affected DNA packaging, progeny yield, and thermostability while maintaining DNA replication, viral cytopathic effect/lateralization, and CAR-dependent binding. Most importantly, the pIX-EGFP label was associated with the capsid due to its direct involvement in viral assembly and conferred upon progeny virions a unique fluorescent property allowing particle detection as hypothesized. The detectability of the particles was demonstrated in a number of applications, including flow cytometry, tracking of infection, and in situ detection after systemic biodistribution with high resolution and sensitivity.
Our system can be used to generate fluorescent adenoviruses at high titers without the need for any additional steps after purification. These fluorescent viral particles will have great utility in the assessment of targeting function both in vitro and in situ following systemic administration. An exciting possibility that has yet to be validated is detecting the biodistribution of our labeled particles in vivo with noninvasive imaging systems. The detectability of adenoviral particles in this sense with our genetic labeling system would rely on a number of parameters, including the mode of detection, the location of the particles, the local concentration of the vector, and most importantly, the biological contrast agent used to label the virus. All of these factors will influence the tissue penetration of the signal and therefore the lower threshold of detection.
It is a great challenge to directly detect nanoscale viral particles with any imaging modality from outside the body. This consideration is especially true for the EGFP label used in this study which is only detectable within a few millimeters deep in tissue [37]. Recently, we were successful in expanding the versatility of our genetic labeling strategy by incorporating other imaging ligands onto pIX, including red fluorescent proteins (mRFP1 and tdimer2 [12]) [38] and Renilla and firefly luciferases [39] (data not shown). These imaging ligands may offer more promise with regard to noninvasive detection of genetically labeled adenoviral particles following biodistribution. Furthermore, other biological contrast agents such as herpes simplex virus thymidine kinase [9,10], gadolinium-based avidin-biotin [40], or proteases [41] may be configured onto pIX to generate labeled particles compatible with clinically relevant PET, MRI, and near-infrared fluorescence imaging, respectively.
While noninvasive imaging of adenoviral vector biodistribution poses formidable challenges, our genetic adenovirus labeling system may have additional impact in the field of adenoviral virotherapy. This therapeutic modality shows promise in eradicating tumors by harnessing the lytic property of adenovirus replication; however, lack of an imaging system to monitor the replication and spread of oncolytic adenoviruses has hampered the development and study of these agents. Our labeling scheme can plausibly be applied to replicative adenoviral agents to detect replication and localization because it conceptually mimics staining of viral structural proteins such as hexon, a standard technique used to assess replication in clinical trials [42,43]. The essence of viral replication would serve as a signal amplification step while the genetic basis of our adenoviral labeling system would allow dynamic monitoring of oncolytic adenoviruses both temporally and spatially. Moreover, if combined with the abovementioned imaging modalities, the system possesses potential for in vivo imaging of adenoviral replication and spread. Likewise, the exact same method can be used to monitor the mechanism of infection in the study of adenoviral pathogenesis.
Our labeling system offers a powerful alternative to conventional methods of adenoviral vector detection. Genetically labeled adenoviruses will have important implications for vector development, adenoviral targeting, and adenovirus biology as well as potential for monitoring oncolytic adenoviruses.
Footnotes
Acknowledgments
We thank Dr. Sven Pettersson for his kind gift of the hCAR transgenic mouse and Dr. Joel Glasgow for his critical review of the manuscript. This work was supported by NIH grants P50 CA83591, RO1 CA83821, RO1 CA93796, RO1 CA94084, RO1 HL67962-3, RO1 DK063615, and DAMD17-03-1-0104, the Susan G. Komen Foundation, and the Medical Scientist Training Program at the University of Alabama at Birmingham.