
Abstract
Multidrug resistance (MDR) mediated by overexpression of MDR1 P-glycoprotein (Pgp) is one of the best characterized barriers to chemotherapy in cancer patients. Furthermore, the protective function of Pgp-mediated efflux of xenobiotics in various organs has a profound effect on the bioavailability of drugs in general. Thus, there is an expanding requirement to noninvasively interrogate Pgp transport activity in vivo. We herein report the Pgp recognition properties of a novel 99mTc(I)-tricarbonyl complex, [99mTc(CO)3(MIBI)3] + (Tc-CO-MIBI). Tc-CO-MIBI showed 60-fold higher accumulation in drug-sensitive KB 3–1 cells compared to colchicine-selected drug-resistant KB 8-5 cells. In KB 8-5 cells, tracer enhancement was observed with the potent MDR modulator LY335979 (EC50 = 62 nM). Similar behavior was observed using drug-sensitive MCF-7 breast adenocarcinoma cells and MCF-7/MDR1 stable transfectants, confirming that Tc-CO-MIBI is specifically excluded by overexpression of MDR1 Pgp. By comparison, net accumulation in control H69 lung tumor cells was 9-fold higher than in MDR-associated protein (MRP1)-expressing H69AR cells, indicating only modest transport by MRP1. Biodistribution analysis following tail vein injection of Tc-CO-MIBI showed delayed liver clearance as well as enhanced brain uptake and retention in mdr1a/1b(−/−) gene deleted mice versus wild-type mice, directly demonstrating that Tc-CO-MIBI is a functional probe of Pgp transport activity in vivo.
Keywords
Introduction
Emergence of multidrug resistance (MDR) is a major obstacle to successful chemotherapy of cancer and is characterized by the failure to respond to a variety of structurally and functionally diverse natural product cytotoxic and xenobiotic agents [1],[2]. MDR1 P-glycoprotein (Pgp), an Mr 170,000 transmembrane glycoprotein, is a well-studied mediator of MDR. When functionally present in cell membranes, Pgp decreases intracellular accumulation of anticancer drugs putatively by acting as an energy-dependent efflux transporter which efficiently pumps drugs out of cells [1–3]. Although the exact molecular mechanism of Pgp-mediated drug resistance is yet to be determined, evidence exists for contributions from mechanisms in addition to drug efflux, including altered membrane permeability resulting in decreased drug influx [4],[5], altered intracellular distribution of drug [6],[7], and modulation of programmed cell death pathways [8],[9].
MDR1 Pgp has been a target for cancer therapy on two fronts. First, reversal of MDR in tumor cells by nontoxic agents that block the transport activity of Pgp has been an important target of pharmaceutical development [3,10–15]. When coadministered with a cytotoxic agent, these inhibitors, known as MDR modulators, enhance net accumulation of cytotoxic compounds within the tumor cells. Second, transgenic expression of the MDR1 gene has been explored for hematopoietic cell protection in the context of cancer chemotherapy [16–18], wherein Pgp could protect hematopoietic progenitor cells from chemotherapy-induced myelotoxicity. Hematopoietic cells transduced via retroviral-mediated transfer of the MDR1 gene have shown preferential survival in vivo after treatment with MDR drugs [18] and recent pilot clinical data support the approach [19]. For applications relevant to the use of new modulators as well as gene therapy in chemotherapeutic protocols, identification of transporter-mediated resistance could guide the choice of chemotherapeutic agents and provide important prognostic information in cancer patients. Thus, noninvasive imaging with a radiolabeled transport substrate serving as a surrogate marker of chemotherapeutic agents may identify those tumors and tissues in which Pgp is expressed and active.
In addition to expression in tumors, MDR1 Pgp normally is present in various tissues including the intestinal epithelium, choroid plexus epithelium, kidney, liver, adrenal, placenta, and capillary endothelial cells of the blood-brain and blood-testis barriers [20],[21]. Studies in mice that are genetically deficient in mdr1a and mdr1b genes confirmed the pivotal role of murine mdr1 in normal absorption and excretion of many commonly used drugs and xenobiotics as well as its key role in regulating cellular and tissue levels of these pharmacological agents [22]. Furthermore, single nucleotide polymorphisms have been identified in the human MDR1 gene that confer 4-fold differences in oral absorption of MDR drugs [23]. Inhibition of Pgp with an MDR modulator could provide an effective means for increasing oral uptake of drugs and reducing drug excretion, resulting in decreased dosing requirements. For example, Pgp modulation is currently being evaluated as a means to improve oral absorption of chemotherapeutics and HIV-1 protease inhibitors [24],[25]. Similarly, the use of MDR modulators could allow drug penetration into Pgp protected sites in the body, such as the brain and selected hematopoietic cells, as has been shown for penetration of protease inhibitors into the central nervous system [25]. Therefore, a noninvasive method of determining Pgp-mediated drug transport activity could be important in predicting oral absorptivity, pharmacokinetics, and individually specific penetrance of MDR drugs into the brain, germ cells, and fetus.

Structures of M-Carbonyl, M-CO-MIBI, and Tc-Sestamibi complexes.
Gamma-emitting compounds exemplified by hexakis(2-methoxyisobutyl isonitrile) 99mTc(I) (Tc-Sestamibi) (Figure 1) and [1,2-bis{bis(2-ethoxyethyl) phosphino} ethane]2O2 99mTc(V) (Tc-Tetrofosmin), widely available radiopharmaceuticals originally designed as myocardial perfusion agents, have been characterized as substrates for MDR1 Pgp [26–32] and may enable scintigraphic analysis of Pgp-mediated transport activity with imaging cameras commonly available in nuclear medicine facilities [33–43]. An attractive alternative approach to Tc-99m-labeling using the organometallic aquaion [99mTc(CO)3 (OH2)3]+ (
Materials and Methods
Preparation of [99mTc(CO)3(MIBI)3]+
Synthesis of the radiolabeled complex [99mTc(CO)3 (MIBI)3] was accomplished with a two-step procedure (Figure 2). First, the [99mTc(CO)3(OH2)3]+ intermediate (I) was prepared by adding 1 mL of 99mTcO4− from a commercial generator (20–80 mCi) to a 10-ml sealed glass vial containing a lyophilized formulation (10 mg NaKtartrate, 10 mg K2B4O710H2O, and 4 mg potassium boranocarbonate) and heating the vial in a boiling water bath for 15 min. In the second step, 0.08 ml of 1 N HCl was added to the [99mTc(CO)3(OH2)3]+ intermediate reaction vial followed by 0.2 ml of a kit matrix (Cardiolite, Du Pont Medical Products Division, N. Billerica, MA) that had been reconstituted with 1.0 ml of water. The resulting solution was heated for 30 min at 75°C. Separation of the radiolabeled complex from radiochemical and chemical impurities was accomplished using a Waters C-18 Sep-Pak cartridge as previously described [46]. Quality control effected by reverse-phase HPLC [C-18 Vydac column (250 × 4.6 mm, 5 μm) with a 0.05-M TEAP/methanol gradient (0–100% MeOH in 30 min)] showed >90% radiochemical purity (tr = 20.2 min). Final concentration of the ethanol vehicle in experimental buffers was #x003C; 0.5%, which has been shown previously to have no effect on the transport kinetics of a 99mTc-complex in cultured cells [46].
Preparation of [Re(CO)3(MIBI)3](NO3)
Synthesis of the corresponding rhenium complex was performed by modification of general methods described previously [47]. (NEt4)2 [Re(CO)3(Br)3] (3.2 mg, 0.0042 mmol) was dissolved in 0.1 ml H2O. Three equivalents of AgNO3 (dissolved in 0.1 ml H2O) were added and the precipitated AgBr was filtered yielding
Cationic 99mTc species were additionally characterized by paper electrophoresis [200 V, 20 V/cm, 1 h; Whatman CHR 1 paper, acetonitrile/0.1 M phosphate buffer (50/50 v/v), pH 7.4]. In addition, relative lipophilicity data were obtained by measuring HPLC capacity factors (k′), defined as k’ = [tr – to]/to (where tr = retention time of test compound and to = time of void volume), employing a PRP-1, 150 × 4.1 mm, 10 μm column in 45:55 CH3CN:0.1 M NH4OAc at a flow rate of 2.0 ml/min. HPLC capacity factors obtained on this isocratic system have demonstrated good correlation with octanol/buffer partition coefficients of a variety of 99mTc-complexes (unpublished data).

Reaction scheme for synthesis of Tc-CO-MIBI.
Cells and Solutions
Monolayers of human epidermoid carcinoma drug-sensitive KB 3-1 (Pgp —) cells and the colchicine-selected KB 8-5 (Pgp+) derivative cell lines were grown as previously described [26,48,49]. Briefly, cells were plated in 100-mm Petri dishes containing seven 25-mm glass coverslips on the bottom and grown to confluence in Dulbecco's modified Eagle's media (DMEM, Gibco, Grand Island, NY) supplemented with l-glutamine (1%), penicillin/streptomycin (0.1%), and heat-inactivated fetal bovine serum (10%) in the presence of 0 and 10 ng/ml colchicine, respectively. Growth conditions for human H69 (MRP1—) and drug-selected H69AR (MRP1+) have been described [49–51]. Parental human MCF-7 (Pgp —) cells were cultured in DMEM (Gibco), l-glutamine (1%), penicillin/streptomycin (0.1%), and fetal bovine serum (10%) in a 5% CO2 incubator at 37°C. MCF-7/MDR1 (Pgp+) cells were derived as described (G. Luker and D. Piwnica-Worms, submitted). Briefly, pGEM3Zf(-)Xba-MDR1.1 was purchased from the ATCC and sequenced to confirm wild-type MDR1. MDR1 was excised with XbaI and subcloned into pBluescript SK (Stratagene, La Jolla, CA). The cDNA for MDR1 was then excised with NotI and BamHI and ligated into corresponding sites of pIRESneo (Clontech, Palo Alta, CA). Cells were transfected with MDR1 or vector using Fugene 6 (Roche, Nutley, NJ). Clones of transfected cells were isolated and maintained in medium containing 1 mg/ml G418.
Stock solutions of GF120918 (gift of Glaxo-Wellcome, Research Triangle Park, NC), LY335979 (gift of Eli Lilly, Indianapolis, IN), PSC 833 (gift of Mallinckrodt, St. Louis, MO), methotrexate, and cis-platin were prepared in dimethyl sulfoxide (DMSO). Final concentration of DMSO in experimental buffers was <0.5% which has been found to have no effect on net uptake of 99mTc-complexes in cultured cells [46]. 99mTc-Sestamibi was prepared from Cardiolite kits as described [46]. All reagents except as indicated were obtained from Sigma, St. Louis, MO.
Control buffers for transport experiments were either DMEM plus bovine calf serum (1%) or a modified Earle's balanced salt solution (MEBSS) containing (mM): 145 Na+, 5.4 K+, 1.2 Ca2+, 0.8 Mg2+, 152 Cl−, 0.8 H2PO4−, 0.8 SO42−, 5.6 dextrose, 4.0 HEPES, and bovine calf serum (1%, vol/vol), pH 7.4 ± 0.05.
Cell Transport Studies
Coverslips with confluent cells were used for studies of cell transport and kinetics as previously described [27]. Cells on coverslips were removed from culture media and washed for 15 sec in control buffer, then immersed in 60-mm glass Pyrex dishes containing 4 ml of loading buffer with drug or vehicle and 5–20 pM Tc-complex (5–9 pmol/mCi; 1–2 μCi/ml). Coverslips with cells were removed at various times, rinsed three times in 25 ml ice-cold isotope-free control buffer for 8 sec each to clear extracellular spaces, and placed in 35-mm plastic Petri dishes. Cells were then extracted in 1% sodium dodecylsulfate with 10 mM sodium borate before protein assay by the method of BCA analysis (Pierce Chemical) using bovine serum albumin as the protein standard. Aliquots of the loading buffer and stock solutions also were obtained for standardizing cellular data with extracellular concentration of Tc-complex. For cells in suspension culture, transport studies were performed as described [52]. All cell extracts, stock solutions, and extracellular buffer samples were assayed for gamma activity in a well-type sodium iodide gamma counter (Cobra II, Packard, Meridian, CT). The absolute concentration of total Tc-complex in solution was determined from the activity of stock solutions and specific activity of technetium, based on equilibrium equations for the Mo/Tc generator [53]. Data are reported as fmol Tc-complex (mg protein)−1 (nMo)−1 as previously described [27], with nMo representing total concentration of Tc-complex (as both 99mTc and 99Tc) in the extracellular buffer.
A concentration-effect curve was generated from a 30-min Tc-CO-MIBI accumulation experiment in the absence or presence of various concentrations of LY335979. For washout experiments, cells on coverslips were equilibrated in loading buffer for 1 h, washed three times in ice-cold control buffer, then incubated in isotope-free control buffer (37°C). Preparations were removed at various times and assayed for retained cell-associated counts as described above.
Western Blots
Pgp and MRP1 were detected in crude, enriched membrane preparations from cell lines using Western blotting with monoclonal antibodies C219 and QCRL1 (Signet, Dedham, MA), respectively [21]. Immune complexes were identified with sheep anti-mouse antibody (1:2,000 dilution) coupled to horseradish peroxidase using the ECL Western blotting detection system (Amersham Life Sciences, Arlington Heights, IL).
Biodistribution Studies
Vertebrate animal procedures were approved by the appropriate institutional review committees. Distributions of Tc-Sestamibi and Tc-CO-MIBI in tissues of parental strain FVB mice (FVB/NTac) and FVB mdr1a/1b(−/−) gene deleted mice (FVB/Tac-Pgy1 tm1 — Pgy2 tm1 N7) (Taconic, Germantown, NY) were determined as previously described [49],[54]. Tc-com-plexes were diluted in saline containing up to 10% ethanol (final concentration 20–40 μCi/ml). Mice were anesthetized by metofane inhalation and injected with 2 μCi (5–9 pmol/mCi) of radiotracer via bolus injection through a tail vein. Animals were sacrificed by cervical dislocation at 5 and 120 min postinjection (n = 2–4 each). Blood samples were obtained by cardiac puncture and tissues harvested rapidly. Gamma activity in organ samples was counted for 1 min, or until two standard deviations of sampling were below 0.5%. Data are expressed as percentage of injected dose per gram of tissue [(tissue μCi) (injected μCi)−1 (g tissue)−1 × 100] or injected dose per organ [(tissue μCi) (injected μCi)−1 × 100].
Imaging Studies
FVB wild-type and FVB mdr1a/1b(−/−) mice were anesthetized with metofane inhalation. Tc-CO-MIBI (500 μCi in 50 μl saline) was injected via a tail vein into mice positioned under a gamma scintillation camera (Siemens Basicam, Siemens Medical Systems, Iselin, NJ; 5 mm pinhole collimator; 20% energy window centered over 140 keV photopeak of Tc-99m). Sequential posterior images of mice were collected for 5–10 min each at 5, 120, and 240 min postinjection of the radiotracer. 125 × 125 or 256 × 256 image matrices with correction for radioactive decay were used on a PC platform and standard region-of-interest (ROI) image analysis software. No corrections were made for scatter or attenuation. Whole body distributions of the Tc-complex are presented with pseudocolor images.
Analysis
All data points for transport assays were determined from preparations obtained from the same culture. The EC50 value for LY335979 was estimated by computer fit (Sigma Plot, SPSS) of a concentration-effect curve of Tc-complex transport inhibition using the following sigmoid equation:

where C is cell content of Tc-complex, Cmax is maximum cell content of Tc-complex, Cmin is minimum cell content of Tc-complex, γ is the slope, D is concentration of MDR modulator, and EC50 represents the half-maximal effective concentration [55].
Data are generally reported as mean ± s.e.m. Pairs were compared by Student's t test. Values of p ≤ .05 were considered significant.
Results
Chemistry
Tc-CO-MIBI was prepared in two steps (Figure 2). The first step involved the quantitative formation of the carbonyl precursor, [99mTc(CO)3(OH2)3]+, by direct carbonylation of pertechnetate in saline using a lyophilized kit formulation containing potassium boranocarbonate, which acts as both a reductant and a single solid source of carbon monoxide. After neutralizing the solution in step 1 with HCl, exchange of the three water ligands with three MIBI ligands was accomplished by adding a portion of a Cardiolite kit matrix to the carbonyl precursor and heating. Tc-CO-MIBI yields based on HPLC using radiometric detection were ca. 90% (tr = 20.2 min,
Identities of
Confirmation of the cationic charge of both 99mTc species at physiologic pH was achieved using paper electrophoresis, where
Validation of Tc-CO-MIBI as a Substrate for MDR1 Pgp: Transport Analysis in KB Cells

Kinetics of Tc-CO-MIBI accumulation in KB tumor cells (KB 3-1, •; KB 8–5, 6). Cells were incubated in buffer for the indicated times. Each point represents the mean of four determinations; bars represent ± s.e.m. when larger than the symbol. Inset: Efflux of Tc-CO-MIBI. Cell-associated activity during 120-min washout period is plotted. Each point represents the mean of four determinations; bars represent ± s.e.m. when larger than the symbol.
To validate the transport of Tc-CO-MIBI by MDR1 Pgp, cell accumulation of the tracer was determined in drug-sensitive KB 3-1 and drug-resistant KB 8-5 cell lines. Western blots of enriched plasma membrane preparations confirmed that KB 3-1 cell lines express no immunodetectable MDR1 Pgp, while KB 8–5 express moderate levels of Pgp [21,51]. We tested the hypothesis that net cellular accumulation of the lipophilic cation Tc-CO-MIBI, like Tc-Sestamibi, would reflect the balance of influx driven by the physiologically negative mitochondrial and plasma membrane potentials [27,46] opposed, when functionally present, by MDR1 Pgp-mediated outward transport. Time-activity curves show large differences in accumulation of Tc-CO-MIBI between KB 3-1 and KB 8-5 cells (Figure 3). Cell-associated counts were normalized to total Tc-CO-MIBI concentrations in the loading buffer and to protein content. Tc-CO-MIBI demonstrated continuous accumulation in nonresistant KB 3-1 cells through 3 hr of incubation [final: 181.0 ± 4.0 fmol (mg protein)−1 (nMo)−1], while in KB 8–5 cells, time-activity curves reached a low plateau within 5 min of initial exposure to Tc-CO-MIBI in the uptake buffer [0.5 ± 0.1 fmol (mg protein)−1 (nMo)−1]. Furthermore, when KB 3–1 cells were preincubated (60 min) in loading buffer containing Tc-CO-MIBI, then transferred to isotope-free control solution, cell-associated activity was still 36.0 ± 2.6 fmol (mg protein)−1 (nMo)−1 after a 120-min washout period (Figure 3, inset). Uptake (30 min) of Tc-CO-MIBI was 47.3 ± 2.3 fmol (mg protein)−1 (nMo)−1 in KB 3-1 cells and 0.8 ± 0.1 fmol (mg protein)−1 (nMo)−1 in KB 8-5 cells, approximately half the values of 104.6 ± 4.1 and 2.8 ± 0.1 fmol (mg protein)−1 (nMo)−1, respectively, obtained for Tc-Sestamibi under the same conditions [51]. The ratio of 30-min accumulation of the radiopharmaceutical in KB 3-1 and KB 8-5 cells was used as a measure of functional MDR1 Pgp, thereby correcting for differences in absolute cell content of each Tc-complex. The ratio of cell accumulation (KB 3-1/KB 8-5 cells) for Tc-CO-MIBI was 59, nearly double the ratio for Tc-Sestamibi.
To further define the interactions of Tc-CO-MIBI with cells, monolayers of confluent KB 3–1 cells were incubated with tracer amounts of the Tc-complex in buffer alone or buffer containing 130 mM K+/20 mM Cl− and 1 μg/ml of the potassium ionophore valinomycin. Under high K+ plus valinomycin conditions, transmembrane electrical potentials of the mitochondrial inner membrane (Δ) and plasma membrane (Em) are depolarized toward zero, eliminating the inward driving force for uptake of hydrophobic cations such as Tc-CO-MIBI or Tc-Sestamibi [46],[56]. The residual net accumulation of a radiopharmaceutical under isoelectric membrane potential is one measure of nonspecific adsorption of hydrophobic cationic complexes to lipid compartments within cells [46],[57]. Prior studies have established that Tc-Sestamibi is a Nernstian probe of membrane potential with minimal adsorptive binding to lipid bilayers [46,58,59]. Thus, in the presence of high K+ and valinomycin, residual uptake of Tc-Sestamibi maps intracellular water space [26,46,58]. For Tc-CO-MIBI, net uptake in KB 3-1 cells (30 min incubation) under isoelectric membrane potential conditions was 17.2 ± 0.9 fmol (mg protein)−1 (nMo)−1, which was a 65% decrease compared to normal nondepolarizing conditions, yet significantly higher than the expected residual uptake into intracellular water space (3.3 fmol (mg protein)−1 (nMo)−1 [26] and unpublished data). These data indicated a partial membrane potential-dependent uptake mechanism with a significant component of nonspecific binding to hydrophobic domains. Partial potential-dependent uptake of Tc-CO-MIBI with some incorporation into lipid membrane compartments was observed in all cells investigated.
To further characterize transport of Tc-CO-MIBI mediated by MDR1 Pgp, accumulation of the complex in KB cells after 60 min incubation was determined as a function of extracellular concentration of Tc-CO-MIBI. At extracellular concentrations of Tc-CO-MIBI between 70 fM and 1 nM, cell content of the Tc-carbonyl complex was constant at 125.2 ± 12.2 fmol (mg protein)−1 (nMo)−1 in KB 3–1 cells and essentially zero in KB 8–5 cells. Thus, no evidence of saturation of transport kinetics was observed over the concentrations analyzed. A similar large capacity for transport of Tc-Sestamibi by MDR1 Pgp has been reported previously over a much greater concentration range with no saturation of transport seen between 7 pM and 10 μM [27]. It should also be noted that when kinetic experiments were performed at 4°C, no uptake of Tc-CO-MIBI in KB 3–1 cells was observed (data not shown), consistent with an energy-dependent translocation mechanism involving the lipid bilayer.
Pharmacological Analysis in KB Cells
To determine if cell content of Tc-CO-MIBI could be increased by inhibition of Pgp, three potent modulators of MDR1 Pgp function: GF120918, a substituted isoquinolinyl acridonecarboxamide [11]; LY335979, a difluorocyclopropyl dibenzosuberane [12]; and PSC 833, a cyclic undecapeptide analogue of cyclosporin A [10], were tested in KB cells. Inhibition of a putative efflux transport of Tc-CO-MIBI would be indicated by an increase in net accumulation of the tracer. For initial screening, cells were incubated for 30 min in uptake buffers in which saturating concentrations of GF120918 (300 nM), LY335979 (1 μM), or PSC 833 (2 μM) had been added. Enhanced accumulation of Tc-CO-MIBI in KB 8-5 cells was observed with all three modulators with little effect on net tracer accumulation in KB 3–1 cells under identical conditions (Figure 4). Uptake levels (30 min) in KB 8–5 cells were 44.9 ± 2.7, 51.2 ± 3.3, and 49.9 ± 1.2 fmol (mg protein)−1 (nMo)−1 for GF120918, LY335979, and PSC 833, respectively, corresponding to 19-, 22-, and 21-fold increases in Tc-CO-MIBI, respectively. Negative control experiments with methotrexate (100 μM) and cis-platin (100 μM) resulted in predictably low cellular accumulation of Tc-CO-MIBI in MDR cells, not differing from control (p > .5), indicating a lack of effect of these non-Pgp drugs on Tc-CO-MIBI transport (Figure 4). A wide range of concentrations of LY335979 was used to determine the apparent potency of this modulator to enhance cell content of Tc-CO-MIBI in KB 8–5 cells (Figure 5). The EC50 value for LY335979 was 62 nM, comparable to that observed with Tc-Sestamibi [60] and Tc-Tetrofosmin [51]. LY335979 had minimal or no effect on uptake in KB 3-1 cells up to a dose of 30 μM.

Effects of MDR modulators and non-MDR drugs on Tc-CO-MIBI cellular accumulation in KB 3–1 and KB 8–5 cells. Cells were incubated in buffer containing Tc-CO-MIBI in the absence or presence of each drug (300 nM GF120918; 1 μM LY335979; 2 μM PSC 833; 100 μM methotrexate; 100 μM cis-platin). Each point represents the mean of four determinations; bars represent ± s.e.m.
Specificity of Tc-CO-MIBI for MDR1 Pgp-Mediated Transport Activity
Because cell lines derived from stepwise selection with MDR drugs may have multiple mechanisms of drug resistance [61], MCF-7 cells were stably transfected with MDR1 (MCF-7/MDR1) without use of MDR drugs (G. Luker and D. Piwnica-Worms, submitted). No immunodetectable Pgp was present in control MCF-7 cells, while MCF-7/MDR1 cells overexpressed Pgp localized predominantly to the plasma membrane. Function of transfected MDR1 Pgp was confirmed with Tc-Sestamibi. Thus, to further validate Tc-CO-MIBI as a transport substrate for MDR1 Pgp, the tracer was screened in MCF-7 and MCF-7/MDR1 cells. Intracellular accumulation of Tc-CO-MIBI was assayed after a 30-min incubation period in control buffer containing the radiotracer using the same method as above. MCF-7 cells showed high Tc-CO-MIBI accumulation [34.9 ± 3.6 fmol (mg protein)−1 (nMo)−1], while MCF-7/MDR1 cells showed very low uptake, indistinguishable from zero. Furthermore, incubating MCF-7/MDR1 cells with a saturating dose of LY335979 (1 μM) enhanced accumulation of Tc-CO-MIBI to approximately half the control levels [13.8 ± 0.7 fmol (mg protein)−1 (nMo)−1].

Concentration-effect of LY335979 on Tc-CO-MIBI cellular accumulation in KB 8–5 cells. Percentage of maximum cellular content of Tc-CO-MIBI is plotted. Each point represents the mean of four determinations; bars represent ± s.e.m. when larger than the symbol.
Biodistribution of Tc-CO-MIBI in FVB wild-type and mdr1a/1b(—/—) mice

Data obtained 5 min and 2 hr post tail vein injection are expressed as both percentage of injected dose of radioactivity per gram tissue and per organ at each respective time point.
Tc-CO-MIBI and MRP1-Mediated Transport Activity
To evaluate collateral transport of Tc-CO-MIBI by MRP1—a related member of the ABC superfamily of membrane transport proteins which confers resistance to an overlapping array of compounds [7]—cell transport assays were performed with the radiotracer using H69 and H69AR cells, where H69AR cells were confirmed to express high levels of MRP1 (in the absence of MDR1 Pgp) [51]. Accumulation data (30 min) showed an H69/H69AR ratio of 8.9, indicating that Tc-CO-MIBI was a modest substrate for MRP1. However, as expected, LY335979 (1 μM) had no enhancing effect on Tc-CO-MIBI accumulation in H69AR cells [19.5 ± 0.3 (−LY) vs. 14.8 ± 0.4 (+LY) fmol (mg protein)−1 (nMo)−1].
Biodistribution and Imaging Studies in mdr1a/1b(−/−) Mice
To characterize the potential utility of Tc-CO-MIBI as a marker of Pgp transport function and modulation in vivo, quantitative pharmacokinetic analysis (at 5 min and 2 hr) and qualitative scintigraphic imaging of mice following intravenous injection of the tracer were performed. Biodistribution data of Tc-CO-MIBI in mdr1a/1b(−/−) mice in comparison to control wild-type FVB mice are given in Table 1, and selected results are graphically depicted in Figure 6 in which corresponding Tc-Sestamibi data are also shown for comparison. For both Tc-Sestamibi and Tc-CO-MIBI, delayed liver washout in mdr1a/1b(−/−) mice versus wild-type mice was observed indicating that the major pathway for liver clearance of both radiotracers was mdr1a/1b Pgp-mediated. Similarly, both agents demonstrated enhanced brain uptake and retention in mdr1a/1b(−/−) mice versus wild-type mice. Although both complexes exhibited a 3-fold enhancement in brain uptake in the gene deleted mice versus control mice, absolute brain uptake was 3.5 times greater with Tc-Sestamibi relative to Tc-CO-MIBI. Blood pharmacokinetics of Tc-CO-MIBI showed no differences in mdr1a/1b(−/−) versus wild-type mice and the data were essentially superimposible with the corresponding Tc-Sestamibi data, signifying that organ differences in Tc-CO-MIBI cannot be attributed to differences in blood content of the tracer. Kidney clearance was slightly decreased in mdr1a/1b(−/−) mice versus controls for both Tc-99m agents (Table 1 and Slapak et al. [60]), indicative of renal clearance involving other mechanisms not expressed in mouse liver in addition to mdr1a/1b Pgp.

Pharmacokinetics of Tc-Sestamibi and Tc-CO-MIBI in blood, liver, and brain of FVB mice. Wild-type (•) andmdr1a/1b(−/−) mice (◦) were administered the Tc-complexes by bolus injection into a lateral tail vein and organs harvested at the indicated times for analysis. Data are expressed as percentage of injected dose of radioactivity per organ at each respective time point. Data points represent the mean of 2–4 determinations each; bars represent ± s.e.m. or the range when larger.
Representative images of whole body uptake of Tc-CO-MIBI at 5 min, 2 hr, and 4 hr postinjection in mdr1a/1b(−/−) and wild-type mice are shown in Figure 7. Initial liver uptakes were similar in mdr1a/1b(−/−) and wild-type mice, while enhanced retention of the Tc-carbonyl complex in liver was easily visualized in the gene deleted mice as compared with control after 2 and 4 hr. ROI analysis of the liver on 2- and 4-hr images showed 91% and 104% of the activity seen on the 5-min image for mdr1a/1b(−/−) mice, respectively, and 77% and 52% for wild-type mice, respectively, each correlating well with the quantitative biodistribution data presented in Table 1.
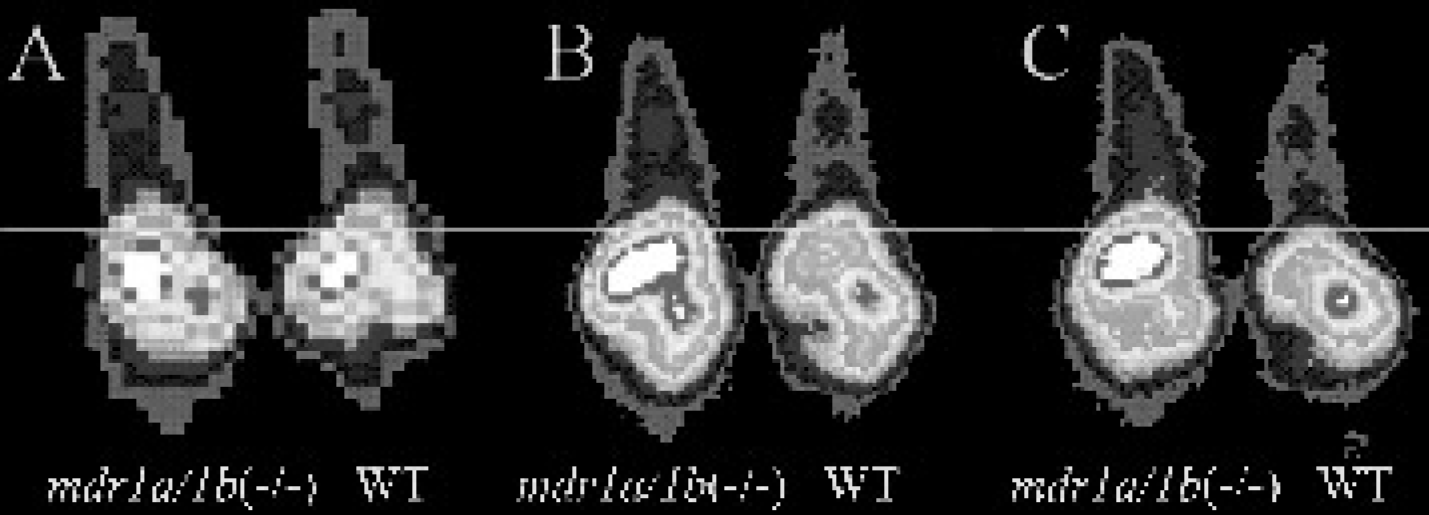
Representative posterior planar images of wild-type (right) and mdr1a/1b(−/−) (left) mice obtained 5 min (A), 120 min (B), and 240 min (C) after intravenous injection of Tc-CO-MIBI. The line references the level of the diaphragm in each image to emphasize Pgp-mediated clearance of activity from the liver and caudal transit of activity over time into the distal intestine in wild-type mice.
Discussion
The radiopharmaceutical application of carbonyl complexes recently became feasible by the development of low-pressure, aqueous syntheses of [M(I)(CO)3(OH2)3]+ complexes (M = Tc, Re), which are convenient starting materials for ligand substitution reactions to obtain new technetium(I) and rhenium(I) complexes containing the facial “M(CO)3” [44]. The characteristics of these carbonyl precursors for the design of radiopharmaceuticals are highly efficient complexation with π-donating ligands, kinetic stability, and small volume of the coordination sphere. The Tc-carbonyl precursor is thus ideally suited to prepare stable Tc(I) monocationic, lipophilic complexes, such as those utilized in cardiology and molecular oncology imaging applications.
In this regard, the tri-substituted [99mTc(CO)3(MIBI)3]+ (
The present study characterized the biochemical pharmacology of Tc-CO-MIBI in human cancer cell lines expressing either MDR1 Pgp or MRP1. The results support the hypothesis that Tc-CO-MIBI is recognized by the MDR1 Pgp. In summary, (1) net cellular accumulation of Tc-CO-MIBI was extremely low in MDR KB 8-5 cells and MCF-cells transfected with MDR1 Pgp resulting in uptake differences of 60-fold or greater between non-Pgp-expressing cells and their Pgp-expressing counterparts; (2) under similar conditions, Tc-CO-MIBI showed almost twice the sensitivity of Tc-Sestamibi for detection of functional Pgp transport activity in vitro [51,62]; (3) known modulators of Pgp increased cell contents of Tc-CO-MIBI in multidrug-resistant cells to levels observed in drug-sensitive cells; (4) the potent and selective MDR modulator LY335979 showed an EC50 value for reversal of Tc-CO-MIBI transport consistent with known EC50 values for reversal of MDR [12] and comparable to those previously reported for Tc-Sestamibi and Tc-Tetrofos-min, other validated radioprobes of Pgp transport [49,51,60]; and (5) Tc-CO-MIBI also appears to be recognized as a transport substrate by MRP1, but to a lesser extent than MDR1 Pgp, since only a 9-fold difference in cellular accumulation was observed with Tc-CO-MIBI between control and MRP1-expressing cells relative to the 60-fold or greater differences observed with cells expressing similar levels of MDR1 Pgp. By comparison, Tc-Sestamibi and Tc-Tetrofosmin show 11-fold and 7-fold differences in these MRP1 expressing cells, respectively [51].
Analysis of the quantitative transport data for Tc-CO-MIBI revealed some interesting trends regarding possible biochemical mechanisms of Pgp. Net cell content of hydrophobic cationic 99mTc-based complexes analogous to Tc-CO-MIBI generally is a function of both passive potential-dependent influx and putative transporter-mediated efflux. For example, Tc-Sestamibi has a well-characterized quantitative membrane potential-dependent response revealed by high K+/valinomycin buffer [46,59] that is equal in magnitude to the tracer content differential between the non-Pgp and MDR1 Pgp-expressing cells. Although Tc-CO-MIBI did show significant membrane potential-dependent uptake properties, it did not respond in as robust a manner as Tc-Sestamibi. In addition, while accumulation of Tc-CO-MIBI in KB 8–5 cells reached a plateau within 5 min, net uptake in KB 3–1 cells continued to increase through 3 hr of incubation with no evidence of equilibrium. Continuous uptake and modestly slow efflux of Tc-CO-MIBI in drug-sensitive cells was consistent with the kinetics observed with a previously synthesized Tc-complex, hexakis(3,4,5-trimethoxyphenylisonitrile)Tc(I), which demonstrated significant adsorption into membranes and cellular redistribution through membrane recycling [63]. In contrast, other cationic, modestly lipophilic Tc-complexes such as Tc-Sestamibi and Tc-Tetrofosmin, achieve steady-state uptake levels after 30–60 min in KB 3–1 cells [27,51,60]. Furthermore, significant cell-associated Tc-CO-MIBI activity remained following a 2-hr washout study. All these data imply that Tc-CO-MIBI may intercalate into lipid bilayers or hydrophobic compartments in a manner that prohibits robust inward translocation in response to imposed transmembrane potentials, yet enables enhanced Pgp-mediated outward translocation by interactions with putative intramembranous domains of the transporter [64]. The membrane adsorption behavior does not appear to be strictly related to the lipophilicity of Tc-CO-MIBI, since the data suggest a more modest lipophilicity relative to Tc-Sestamibi; however, the Tc-tricarbonyl face is relatively non-polar and we hypothesize that it may be responsible for binding to hydrophobic domains.
The observation that uptake of Tc-CO-MIBI is so low (< 1 fmol (mg protein)−1 (nMo)−1) in modestly Pgp-expressing KB 8–5 cells and indistinguishable from zero in high Pgp-expressing MCF-7/MDR1 cells raises some skepticism regarding the putative efflux transport mechanism of Pgp. If passive influx is unaffected, Tc-CO-MIBI should enter Pgp-expressing cells putatively with a portion binding to hydrophobic domains in the membrane and a significant portion being driven into the mitochondria by membrane potentials. It is difficult to envision that Pgp could pump out essentially all of this potential-driven as well as nonspecific compartmentalized activity in less than 5 min after incubation. A more plausible explanation would characterize Pgp as acting as an energy-dependent membrane permeability barrier inhibiting drug influx. A number of investigations also support this proposed mechanism [4],[5].
Mice have two isoforms of Pgp (mdr1a and mdr1b) which confer MDR [65]. Drug-transporting mdr1a Pgp is expressed in capillary endothelial cells of the brain wherein the protein is a major component of the blood-brain barrier [66]. Here, Pgp limits entry of a variety of amphipathic compounds into the central nervous system [65]. Drug-transporting Pgp isoforms are also expressed along the biliary canalicular surface of hepatocytes wherein the transporters function to secrete substrates into the bile [65]. In this regard, mdr1a/1b(−/−) gene deleted mice, which have no drug-transporting Pgp, are a robust model for evaluation of candidate MDR agents by observing hepatobiliary clearance and brain uptake of candidate radiotracers [49]. Biodistribution and imaging results in wild-type and mdr1a/1b(−/−) mice demonstrated the utility of Tc-CO-MIBI as a noninvasive imaging probe of Pgp-mediated transport activity in the liver and brain. Indeed, in the liver and brain, Tc-CO-MIBI was comparable to Tc-Sestamibi in differentially mapping Pgp expression in each tissue. However, in the brain, the absolute level of tracer was 3.5-fold higher for Tc-Sestamibi compared to Tc-CO-MIBI, resulting in relatively superior rendering of the brain with Tc-Sestamibi when using planar scintigraphy. The relative utilities of each tracer in vivo using tomographic techniques applied to brain and tumor models remain to be investigated.
In conclusion, Tc-CO-MIBI may be useful as a surrogate marker of transporter-mediated MDR in tumors and tissues in vivo. Effective inhibition following administration of highly potent and specific modulators can be detected in vitro and in vivo may provide a means for specific detection of MDR1 Pgp [35],[36]. Tc-CO-MIBI has several Pgp targeting properties superior to those of Tc-Sestamibi in vitro, but the tracers appear largely comparable in vivo. Thus, pharmacokinetic mapping of Pgp with these Tc-complexes potentially can be used to (1) guide therapy for patients treated with cytotoxic drugs of the MDR phenotype; (2) evaluate the potential of using chemoprotective gene therapy; and (3) aid in drug development by imaging pharmacokinetics and bioavailability before and after administration of Pgp-selective modulators.
Footnotes
Acknowledgments
We thank Delynn Silvestros for assistance with image analysis. This work was supported by grants from the U.S. Department of Energy (DE-FG02-94ER61885), U.S. National Institutes of Health (P20 CA86251), Mallinckrodt, Inc. and a Mallinckrodt Institute of Radiology Summer Research Fellowship to H.M.B.