
Abstract
The design of near-infrared fluorescent (NIRF) probes that are activated by specific proteases has, for the first time, allowed enzyme activity to be imaged in vivo. In the current study, we report on a method of imaging enzyme activity using two fluorescent probes that, together, provide improved quantitation of enzymatic activity. The method employs two chemically similar probes that differ in their degradability by cathepsin B. One probe consists of the NIRF dye Cy5.5 attached to a particulate carrier, a crosslinked iron oxide nanoparticle (CLIO), through cathepsin B cleavable
Keywords
Introduction
Imaging enzymes in tissues using near-infrared fluorescent (NIRF) probes may permit the characterization of many benign and malignant pathologies based on the presence of specific enzymes. Previous NIRF probes consisted of a high-molecular-weight carrier molecule, known as polyethylene graft copolymer (PGC), to which fluorochrome-derivatized peptides have been attached. For example, NIRF probes have been synthesized using peptides that serve as substrates for cathepsin D [1], [2] or matrix metalloproteases [3] and have been used to visualize enzyme activity from outside living animals. Enzymically activated NIRF probes can also be synthesized by the attachment of fluorochrome-derivatized peptides to a high-molecular-weight carrier, which consists of a crosslinked iron oxide nanoparticle (CLIO), as described below. However, with current forms of NIRF imaging technology using a single optical probe, the signal obtained from cells or tissues reflects a sum of both accumulation and activation of the probe.
Here we describe an improved approach for quantifying the activity of proteases in tissues by optical imaging, which provides information on enzyme activity in tissues independent of the amount of probe present. We term this technique “dual wavelength ratio imaging” of enzyme activity, the principle of which is outlined in Figure 1. Dual wavelength ratio imaging involves the synthesis of two chemically similar macromolecular conjugates, one degradable and one nondegradable, each labeled with a different fluorescent dye. Here, we coupled the NIRF dye Cy5.5 to a macromolecular carrier consisting of a long circulating crosslinked iron oxide (CLIO) nanoparticle through a cathepsin B sensitive spacer of

Principle of “dual wavelength ratio imaging” of enzyme activity. The injectate consists of an enzyme-activatable probe with a cleavable peptide linker and a nonactivatable probe. The fluorescence from the two probes in the quenched state gives an arbitrary ratio of Cy5.5 to Cy3.5, e.g. 1:1. When the probes reach an environment where certain target enzymes are active, the peptide linker of the activatable probe gets cleaved, resulting in the dequenching of the attached dye and an increase of its fluorescence. The ratio of Cy5.5 to Cy3.5 increases, e.g., 5:1.
Materials and Methods
Strategy for the Synthesis of Dye Peptide Nanoparticle Conjugates
Figure 2A shows the scheme used to synthesize the enzyme-activatable nanoparticle denoted Cy5.5–R4–SC–CLIO and nondegradable nanoparticle denoted Cy3.5–r4–SC–CLIO. To synthesize these probes, amino-CLIO was reacted with succinimidyl iodoacetate (SIA) and then with either the peptide R4 (R-R-R-R-G-C) or r4 (r-r-r-r-G-C), to yield arginyl peptide conjugates denoted R4–SC–CLIO or r4–SC–CLIO, respectively. SC stands for the thioether linkage between the sulfhydryl group of the cysteine and the carbon of iodoacetic acid. The designation R or r for arginine indicates the L or D steric configuration, respectively.
The two nanoparticles produced, Cy5.5–R4–SC–CLIO and Cy3.5–r4–SC–CLIO, are chemically similar so that their internalization and intracellular processing are expected to be similar. The differences between the probes are (i) the amino acid configuration (
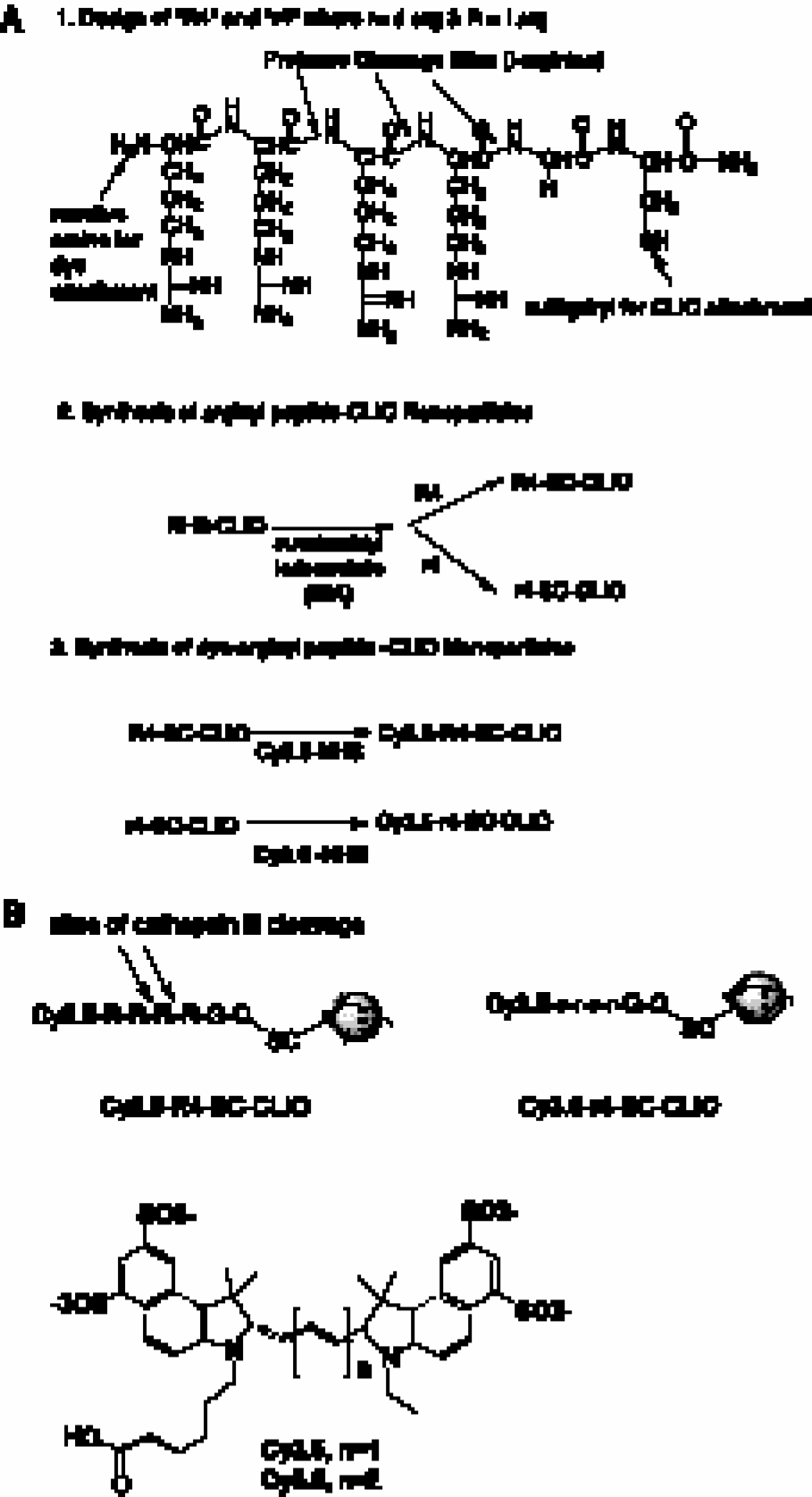
(A) Synthesis of nanoparticles Cy5.5–R4–SC–CLIO and Cy3.5–r4–SC–CLIO. (B) Cleavage of the Cy5.5–R4–SC–CLIO nanoparticle probe and structures of fluorescent dyes.
Physical Properties of Cy3.5–arginyl–CLIO and Cy5.5–arginyl–CLIO Conjugates

Synthesis of Aminated CLIO
A dextran-coated monodispersed iron oxide nanoparticle (MION) colloid was prepared, crosslinked with epichlorohydrin, and reacted with ammonia [4], [5]. It consists of a core of superparamagnetic iron oxide and a crosslinked coating of dextran with amino groups.
Peptide Synthesis
Peptides were synthesized on an automatic synthesizer (PS3, Rainin, Woburn, MA) at a 0.1-mmol scale using Fmoc chemistry with HBTU and HOBT. They were cleaved from Rink amide HBHA resin (Novabiochem, San Diego, CA) with 5 ml TFA/thioanisole/ethanedithiol/anisole (90:5:3:2) and purified by C18 reversed-phase chromatography. Peptides were the
Synthesis of R4–SC–CLIO or r4–SC–CLIO
To aminated CLIO (45 mg, 3 ml) was added 1 ml 0.1 M Na2HPO4 and 1 ml 150 mM succinimidyl-iodoacetate in DMSO. The reaction was allowed to sit for 1 hr at room temperature and the addition of succinimidyl-iodoacetate was repeated. Iodoacetyl-CLIO was separated from iodoacetic acid using a Sephadex G-25 column equilibrated with 0.025 M citrate pH 6.5 run in the cold. The void volume, 10 ml, was split. To 5 ml was added 6–7 mg of R4 or r4 in 0.6 ml of citrate buffer, and the reaction was incubated overnight at room temperature. Unreacted peptide was removed by dialysis against 3 L of 0.025 M citrate pH 8.2 using a 14-kDa cutoff membranes.
Synthesis of Nanoparticle Probes Cy5.5–R4–SC–CLIO or Cy3.5–r4–SC–CLIO
To attach Cy5.5, 200 μL of 1 M NaHCO3 (pH 8.3) was added to 2 ml R4–SC–CLIO (7.2 mg Fe); 440 μl were then added to five tubes of Cy5.5. After 2 hr at room temperature, the tubes were pooled and 220 μl of 1.5 M hydroxylamine was added. The mixture was allowed to sit for an additional 2 hr at room temperature and was separated using PD-10 columns run in a spin separation mode. Columns were washed with 0.025 M citrate pH 8.2 and were spun dry. A 1-ml sample was added to columns that were spun at 2500 × g for 7 min. A similar procedure was followed for the synthesis of Cy3.5–r4–SC–CLIO.
Physical Properties of Nanoparticle Probes
The number of peptides per CLIO was determined as the number of reactive primary amino groups using fluorescamine (Udenfriend, 1972 no. 17 [6]). To accomplish this, 5–50 μl of either r4–SC–CLIO or R4–SC–CLIO or various amounts of the standard peptide (R4) was diluted into 500 μl of 0.1 M borate, pH 8.5. Weight of R4 was determined by amino acid analysis. Fluorescamine (250 μl; 4 mg in 20 ml acetyl nitrile) was added, and the mixture was allowed to stand overnight at room temperature. Trypsin (50 μl; 1 mg/ml in 0.1 M phosphate) was added, and samples were allowed to stand for 2 hr at room temperature. The fluorescence from fluorescamine, ex 390 and em 475, was measured after separating the iron oxide from low molecular weight materials by ultrafiltration [4]. Iron concentration was determined spectrophotometricly [5]. The peptide to crystal ratio was obtained from concentrations of peptide and iron, assuming 2064 iron atoms per crystal [7].
Spectra of conjugates were taken using a Hitachi 3500 spectrophotometer. For Cy5.5–R4–SC–CLIO, the number of dyes per crystal attached was taken from the absorption at 675 nm, and an extinction coefficient of 250,000 M−1 cm−1 and assuming 2064 iron atoms per crystal. For Cy3.5–r4–SC–CLIO, adsorption at 581 nm and an extinction coefficient of 150,000 were used (Amersham-Pharmacia, product information).
The R1 and R2 relaxivities were measured using a 0.47-T tabletop spectrometer as described [4], [5]. Size was determined by laser light scattering using a Coulter N4 particle size analyzer.
Activation of Probes by Proteases
Activation of Cy5.5–R4–SC–CLIO and Cy3.5–r4–SC–CLIO by the protease cathepsin B in vitro was determined in a microtiter plate in 1 mM DTT 0.01 M phosphate buffer, pH 6.0. Cy5.5 and Cy3.5 probes were added in a concentration of approximately 25 μg Fe/ml and cathepsin B in a concentration of 1.6 μg/ml. The plate was incubated at room temperature for various times up to 60 min, and Cy5.5 and Cy3.5 fluorescence was determined (ex 645, em 695, and ex 530, em 595, respectively) using a fluorescent microtiter plate reader (SpectraMAXGeminiXS, Molecular Devices, Sunnyvale, CA).
To determine the ability of macrophages to activate the probes, macrophages were isolated from the peritoneum of C57BL/6 mice 4 days after intraperitoneal injection of 2 ml thioglycollate medium (Difco, Detroit, MI). Macrophages were preincubated alone or in the presence of the membrane-permeable selective cathepsin B inhibitor CA-074 Me (10 μM; Peptide Institute, Osaka, Japan) or E-64-d, a membrane permeable broad band inhibitor of cathepsin B, H, and L (Peptide Institute) in RPMI 1640 (Cellgro; Mediatech, Washington, DC). Media was supplemented with 10% fetal bovine serum (Cellgro). Cells were incubated for 2 hr at 37°C in a humidified atmosphere. Subsequently, 100 μg/ml of each probe was added to the cells and 10 μg/ml to vials without cells. After 6 hr of incubation, macrophages were washed 3 × in Hanks' balanced salt solution (Cellgro) and placed in varying concentrations into microtiter plates. Cells were then lysed with 1% Triton-X (Sigma, St. Louis, MO), and Cy5.5 and Cy3.5 fluorescence was determined with the fluorescence plate reader within 5 min of lysis (SpectraMAXGeminiXS).
Aliquots of macrophages were taken after 4 hr of incubation with the probes, and images were acquired using a CCD camera (Photometrics, Tucson, AZ) connected to a Zeiss Axiovert 100TV microscope (Wetzlar, Germany; 63 × phase-contrast objective). Cy5.5 to Cy3.5 ratio images were calculated using IPLab software (Signal Analytics, Vienna, VA) on a Power Macintosh G3 computer.
Optical Imaging of Tissues
C57CL/6 mice (National Cancer Institute, Bethesda, MD) were injected with a mixture of 60 μg of Cy5.5 probe and 120 μg of Cy3.5 probe in a total volume of 150 μl by tail vein. After 24 hr, animals were sacrificed, and blood samples were taken. The animals were then perfused with 35 ml of NaCl containing 20 U heparin/ml and dissected. Organs (approximately 200 mg each) were placed in 96-well flat bottom microtiter plates (Costar, Corning, NY). Optical imaging of the plates was performed using a whole mouse imaging system, a modification of a commercially available chemiluminescence system, as described [8]. Cy5.5 fluorescence was obtained with an excitation bandpass filter at 630 ± 10 nm (630RDF30; Omega Optical, Brattleboro, VT) and emission bandpass filter at 700 ± 20 nm (700DF40; Omega Optical). Cy3.5 fluorescence was obtained with an excitation bandpass filter at 520 ± 10 (520AF20; Omega Optical) and emission bandpass filter at 600 ± 20 (600AF40; Omega Optical). Images were acquired using a peltier-cooled CCD with 800 × 600-pixel resolution (SenSys; Photometrics). Acquisition settings were aperture 1.2, zoom 20 mm, and acquisition times 30 sec (Cy5.5) and 10 sec (Cy3.5). To correct for the inhomogeneity of the light source, the intensity of both Cy5.5 and Cy3.5 fluorescent images was divided by the light intensity obtained without the emission filter. To quantitate signal intensity of tissues, circular regions of interest of two-thirds of the total well diameter were taken from the center of each well. Kodak Digital Science 1D Software (Eastman Kodak, Rochester, NY) was used for acquisition and data analysis and IPLab software (Signal Analytics) for the creation of ratio images.
Results
As shown in Table 1, Cy3.5–r4–SC–CLIO and Cy5.5–R4–SC–CLIO have similar sizes and magnetic properties. They have similar surfaces, consisting of crosslinked dextran with similar numbers of arginyl peptides attached. The probes are also similar in the nature of the dye attached because Cy3.5 and Cy5.5 have very similar structures (Figure 2).
The absorption spectra of Cy3.5–r4–SC–CLIO, Cy5.5–R4–SC–CLIO, and amine-CLIO are shown in Figure 3. The attachment of the dyes to the iron oxide core slightly shifted their absorption maxima: For Cy3.5, the maximum of 581 nm shifted to 586 nm, while for Cy5.5, the maximum of 675 nm shifted to 680 nm. Also shown is the characteristic absorption of iron oxide, which is pronounced in the ultraviolet region of the absorption spectrum, but which is extremely small in the near-infrared absorbing region.

Absorption spectra of Cy5.5–R4–SC–CLIO, Cy3.5–r4–SC–CLIO, and amino–CLIO.
Figure 4 shows the effect of treating Cy5.5–R4–SC–CLIO and Cy3.5–r4–SC–CLIO with cathepsin B. Incubation with cathepsin B increased the NIRF of Cy5.5–R4–SC–CLIO by cleaving the basic arginine residues in the
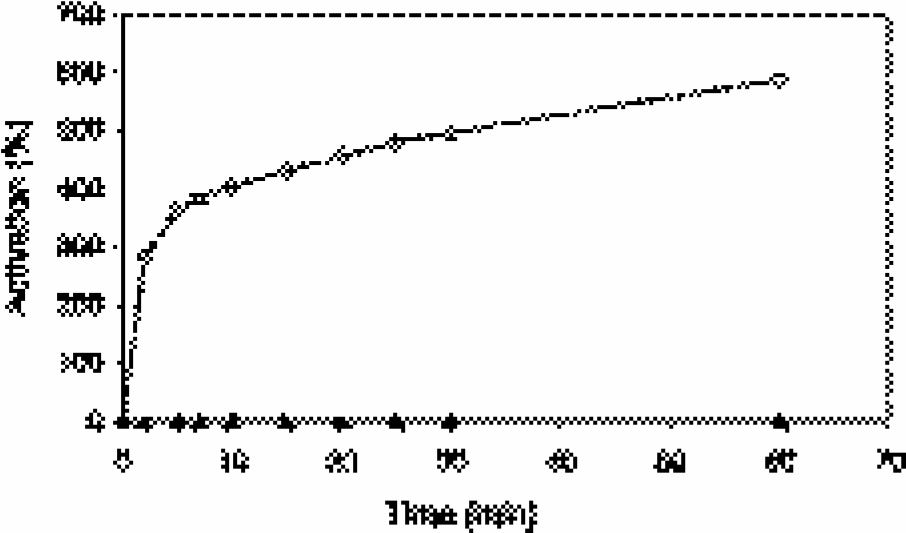
Activation of Cy5.5–R4–SC–CLIO (squares) and Cy3.5–r4–SC–CLIO (triangles) by incubation with cathepsin B (1.6 mg/ml).
To further investigate the combined use of these two nanoparticle probes, a mixture of Cy5.5–R4–SC–CLIO and Cy3.5–r4–SC–CLIO was incubated with different numbers of macrophages (constant volume). Incubations were performed in the presence or absence of a cathepsin B specific inhibitor (CA-074 Me) and in the presence or absence of a broad spectrum inhibitor of cathepsins B, H, L, and calpain (E-64-d). Figure 5A shows the fluorescence intensities (AU) from both Cy5.5 and Cy3.5 channels, while Figure 5B shows the resulting ratios of Cy5.5 to Cy3.5 fluorescence. As shown in Figure 5A, increasing numbers of cells were associated with an increase in fluorescence for both fluorophores, indicating that both were being internalized by macrophages. However, Cy5.5 fluorescence was sensitive to the presence of cathepsin inhibitors, while Cy3.5 fluorescence was not. This indicates that macrophages are activating Cy5.5–R4–SC–CLIO, and not Cy3.5–r4–CLIO, in a similar manner to that seen when nanoparticles were incubated with pure enzyme (see Figure 4). Furthermore, the activation by macrophages is due to cathepsin B, as indicated by the sensitivity of Cy5.5 fluorescence to the presence of CA-074 Me. Finally, the ratio of Cy5.5 to Cy3.5 fluorescence (Figure 5B) was identical for vastly different numbers of cells and reflected the presence or absence of cathepsin activity. Thus, the dual wavelength imaging method can differentiate between high uptake of nanoparticles, obtained with increasing numbers of macrophages, and active cathepsin B in the environment of the nanoparticle.
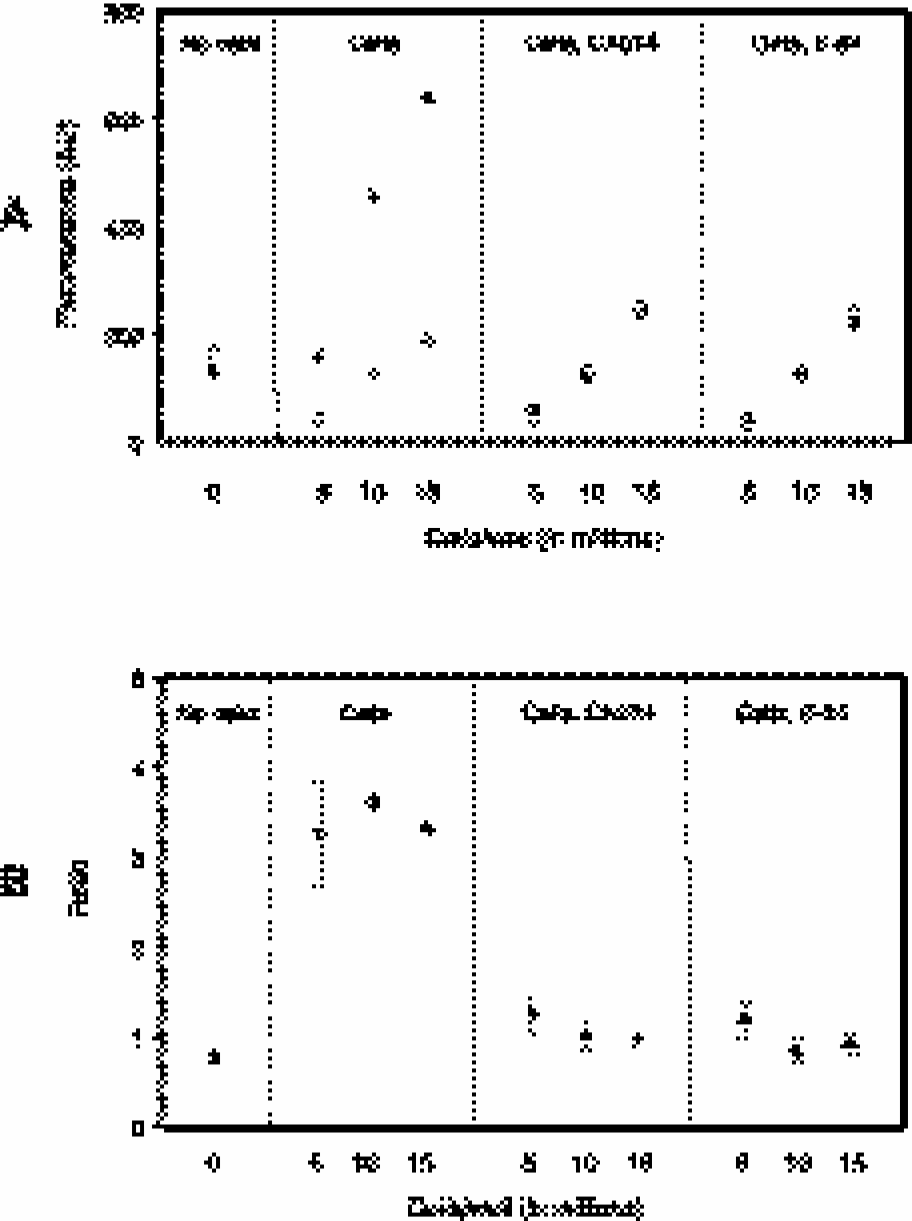
(A) Fluorescence intensities of macrophages as determined with a fluorescence plate reader in the Cy5.5 channel (filled symbols) and Cy3.5 channel (open symbols) after incubation with a mixture of Cy5.5–R4–SC–CLIO and Cy3.5–r4–SC–CLIO. Macrophages were incubated in the presence or absence of a selective cathepsin B inhibitor (CA-074 Me) and an inhibitor with broad specificity (E-64-d). The number of cells per volume was varied (5 × 106, 10 × 106, and 15 × 106 cells/100 μl). Standard deviations were obtained but were too small to be seen graphically (<15% for all values). (B) Fluorescence ratios of Cy5.5 to Cy3.5 resulting from the data in (A). Note the similar ratio values despite varying cell numbers.
Figure 6 shows fluorescence microscopy studies of macrophages incubated with Cy5.5–R4–SC–CLIO and Cy3.5–r4–CLIO for 4 hr. Cy5.5 fluorescence is shown in Figure 6A. The activation of the Cy5.5 probe is readily detectable as punctate regions of high signal intensity in the cytoplasm consistent with the localization of this probe in endosomes. Cy3.5 fluorescence was low because of the quenched state of the probe, however, it was well detectable using individual window and level settings (image not shown) and appeared to be colocalized with the image in the Cy5.5 channel. Using identical window and level settings for Figure 6A and B, Figure 6B appears black. Figure 6C shows a color-coded ratio image resulting from division of Figure 6A and B. Regions of high signal intensity correspond roughly to that obtained with the Cy5.5 image (Figure 6A). Hence, the dual wavelength method can be used to determine the presence of cathepsin B in suspensions of cells or by microscopy.

Microscopy of murine peritoneal macrophages after incubation with a mixture of Cy5.5–R4–SC–CLIO and Cy3.5–r4–SC–CLIO (4 hr, ×63 objective, identical window and level settings). (A) Cy5.5 channel. (B) Cy3.5 channel. (C) Cy5.5 to Cy3.5 ratio image.
We next injected mice with a mixture of Cy3.5–r4–SC–CLIO and Cy5.5–R4–SC–CLIO and obtained fluorescence intensities of both fluorochromes from selected tissues. Figure 7A shows the NIRF fluorescence from the Cy5.5 channel. Liver and spleen had the highest signal intensity, compared with other tissues. Figure 7B shows the fluorescence from the Cy3.5 channel. Figure 7C presents a color-coded image obtained by creating a ratio of Cy5.5 to Cy3.5 fluorescence from Figure 7A and B. Again, liver and spleen had the highest levels of activity, but this activity was now corrected for the high uptake of nanoparticles by these tissues.
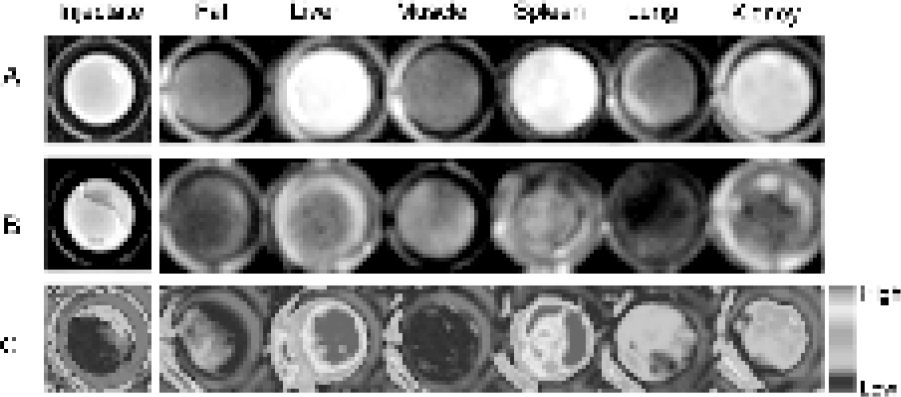
Fluorescent imaging of mouse tissues after injection of a mixture of Cy5.5–R4–SC–CLIO and Cy3.5–r4–SC–CLIO. Tissues were excised 24 hr post injection. (A) Cy5.5 channel at 700 nm emission. (B) Cy3.5 channel at 600 nm emission. (C) Cy5.5 to Cy3.5 ratio image.
To obtain a more quantitative estimate of tissue fluorescence, two additional mice (n = 3) were injected with both probes, and the tissue fluorescence was quantified for all three animals, using regions of interest that were taken over the microtiter wells. The injectate had a ratio of Cy5.5 to Cy3.5 fluorescence of 8.9 ± 0.45. Ratios obtained for tissues were 59.4 ± 1.9 (liver), 17.0 ± 2.5 (muscle), 26.6 ± 2.4 (spleen), 25.5 ± 4.5 (lung), and 25.0 ± 1.9 (kidney). The increase in the ratio of Cy5.5 to Cy3.5 fluorescence over that of the injectate indicates the activation of the Cy5.5 probe by cathepsin B and is consistent with the broad biodistribution of cathepsin B.
Discussion
To our knowledge, this is the first instance where two fluorescent probes of differing degradability have been used as a mixture to determine the activity of an enzyme based on the ratio of fluorescence intensities. We refer to this as “dual wavelength ratio imaging” of enzyme activity.
The uptake and activation of the probes was characterized in vitro by incubating a mixture of Cy5.5–R4–SC–CLIO and Cy3.5–r4–SC–CLIO with macrophages (Figure 5). We observed that the ratio of Cy5.5 to Cy3.5 fluorescence increased fourfold over the ratio of fluorescence that characterized the mixture of probes administered to the cells. Both the selective cathepsin B inhibitor CA-074 Me and broad spectrum cathepsin inhibitor E-64-d Me decreased the ratio of Cy5.5 to Cy3.5 fluorescence to that of the starting probe mixture. Hence, it appears that cathepsin B activity is responsible for the increase in the ratio of Cy5.5 to Cy3.5 fluorescence.
An important observation is that when the number of the macrophages incubated with the probe mixture was varied in vitro, the fluorescence in both the Cy5.5 and Cy3.5 channels varied similarly, whereas the ratio of Cy5.5 to Cy3.5 fluorescence remained unchanged (Figure 5). The association of higher levels of Cy5.5 fluorescence, with increasing cell numbers, was interpretable as higher levels of probe internalization at constant levels of enzyme activity per cell, by virtue of data on the uptake of the Cy3.5 probe. These results show that the dual wavelength method corrects for differences in accumulation and represent an important advance towards a better quantification of enzyme activity.
When a Cy5.5–R4–SC–CLIO and Cy3.5–r4–SC–CLIO mixture was injected, all tissues showed higher ratios of Cy5.5 to Cy3.5 fluorescence than the injectate (see Figure 7, bottom row, fluorescence ratios), consistent with the broad distribution of cathepsin B in rodent tissues, see below. Liver, with a high concentration of macrophages (Kupffer cells), showed the highest ratio of Cy5.5 to Cy3.5 fluorescence.
Use of the CLIO nanoparticle as a macromolecular carrier is especially suitable for probes designed to measure cathepsin B because of the high levels of cathepsin B in macrophages and the ability of macrophages to accumulate iron oxide nanoparticles. Cathepsin B has a broad distribution in cells and tissues [9], with particularly high concentrations in macrophages [10–12]. Cathepsin B is elevated in macrophages when they are stimulated with g-interferon or thioglycollate medium [10–13]. Cathepsin B also plays a role as a prognostic marker in breast cancer [14] and small cell lung carcinomas [15]. Magnetic nanoparticles accumulate in macrophages after intravenous injection [16] or when incubated with macrophages in vitro [17].
The dual wavelength ratio imaging method described here offers several avenues for improvement and refinement. First, as additional NIRF fluorochromes are synthesized, it may be possible to obtain pairs of chemically similar fluorochromes with spectral characteristics further shifted to the infrared region of the spectrum. Second, as the cost of NIRF probes decreases, it may be possible to react excess fluorochrome with the peptide on the solid phase and thus achieve a ratio of one fluorochrome per peptide. Here, we reacted the N-hydroxysuccinimide ester of Cy5.5 and Cy3.5 with the arginyl peptide nanoparticles. The result was nanoparticle probes where a minority of peptides carried a fluorochrome (Table 1). Another implication of the method is the potential to utilize peptides for a variety of proteases other than cathepsin B. Attachment of peptides to the macromolecular carrier PGC has been used to synthesize probes that are activated by cathepsin D and matrix metalloproteases. Finally, though we have used the CLIO magnetic nanoparticle simply as a macromolecular carrier for fluorochrome-derivatized peptides, the core of magnetic iron oxide can serve as an MR contrast agent. The feasibility of using Cy5.5–R4–SC–CLIO as a combined optical and MR contrast agent has been demonstrated [18]. Hence, it may be possible to use the dual wavelength ratio method described here to obtain highly accurate information about the activity of specific enzymes and to obtain information on the position of nanoparticles from their effects on T2-weighted MR images.
For the dual wavelength ratio imaging method to be used clinically, pharmaceutically acceptable versions of optical probes will have to be developed. Since dextran-coated iron oxides have long been used clinically, first as iron supplements and then as MR contrast agents, there appears to be nothing in the design of the probes described here that would preclude their clinical use. In addition, the dual wavelength ratio imaging method will require the development of appropriate instrumentation. A NIRF probe has been used to image human breast cancer in vivo [19], and improved instrumentation, including tomographic versions of NIRF imaging, are being rapidly developed [20], [21].
In conclusion, we presented a new strategy for improved enzyme quantification by optical imaging. We demonstrated the feasibility of the “dual wavelength ratio imaging” method and envision that our approach can be widely used for the design of new optical reporter molecules.
Footnotes
Acknowledgments
This work was supported by grants NIH P50 CA86355, P01-CA69246, R33 CA88365, and R24 CA 92782. The authors would like to thank U. Mahmood and V. Ntziachristos for helpful discussions and K. Marten, J. Allport-Anderson, A. Moore, and A. Petrovsky for technical assistance or advice.
M.F.K. was supported by a fellowship from the German Research Foundation (Deutsche Forschungsgemeinschaft).