
Abstract
The gene encoding glial fibrillary acidic protein (GFAP) is down-regulated 24 hr after reversible brain ischemia, such as with a middle cerebral artery occlusion (MCAO). The in vivo imaging of decreased GFAP gene expression in cerebral ischemia was examined in the present studies using a targeted peptide nucleic acid (PNA), which was labeled with 111In, and which hybridized to nucleotides 20–37 of the rat GFAP mRNA. The PNA was monobiotinylated, and was attached to a monoclonal antibody (MAb) to the transferrin receptor (TfR) via a biotin–streptavidin linkage. The TfR MAb enables trans-membrane transport of the PNA antisense radiopharmaceutical from blood to the cytosol of brain cells. The decreased GFAP gene expression at 24 hr after a 1-hr reversible MCAO was confirmed by immunocytochemistry. The [111In]-labeled PNA–MAb conjugate was administered intravenously to anesthetized rats at 24 hr after the 1-hr reversible MCAO, and the brain uptake of the targeted antisense imaging agent was decreased relative to brain regions outside of the infarct zone. These studies provide evidence that decreased expression of a target gene in brain can be imaged in vivo with a sequence-specific PNA, provided the antisense radiopharmaceutical is delivered across cell membranes with a receptor-specific targeting agent.
Introduction
Gene expression is imaged in vivo with small molecules following the administration of an exogenous reporter gene [1–3]. The imaging of endogenous genes of known function is also possible with small molecules that are trapped in the organ by the protein expressed by the target gene [4]. However, the function of many disease-related genes is not known, although the nucleotide sequence is known. Imaging endogenous gene expression in vivo is possible with sequence-specific antisense radiopharmaceuticals [5–7]. However, antisense agents cross cell membranes poorly and the limiting factor in the development of antisense radiopharmaceuticals is trans-membrane targeting [8], apart from other important issues related to metabolic stability, potential toxicity, and the chemistry of radiolabeling of the antisense agent [5–7]. Prior work focused on the development of targeted antisense radiopharmaceuticals used peptide nucleic acids (PNAs) labeled with 125-Iodine [9,10]. With [125I]-PNA antisense radiopharmaceuticals, it was possible to image increased target gene expression in either experimental brain cancer [9], or in a transgenic mouse model of an inherited disease [10], providing the PNA was reformulated with transmembrane targeting technology. In the absence of targeting technology, no gene expression in the brain could be imaged in vivo [9,10], because the PNA did not cross the brain vascular endothelial wall, which forms the blood–brain barrier (BBB) in vivo. However, gene expression could be imaged in vivo if the PNA antisense radiopharmaceutical was conjugated to a trans-membrane delivery system, such as a receptor-specific peptidomimetic monoclonal antibody (MAb) that targeted the transferrin receptor (TfR). The PNA was conjugated to the targeting MAb using avidin–biotin technology. The PNA is monobiotinylated, which enables high affinity attachment of the PNA to a conjugate of streptavidin (SA) and the TfR MAb (Figure 1A).
Targeted PNA antisense radiopharmaceuticals that are radiolabeled with 125-Iodine are not the ideal agents for imaging gene expression in vivo, owing to the rapid conversion of the [125I]-radiopharmaceutical into low molecular weight radiolabeled metabolites such as [125I]-tyrosine. Following degradation of the radiopharmaceutical, the [125I]-tyrosine is released to blood, followed by transport into cells via a plasma membrane neutral amino acid transport system. In contrast, the metabolites generated from the degradation of radiopharmaceuticals labeled with [111In] are not released to the blood [11]. Prior work with peptide radiopharmaceuticals showed that the background uptake was decreased 10-fold with the use of a radiopharmaceutical labeled with 111-Indium, as compared to 125-Iodine [12].
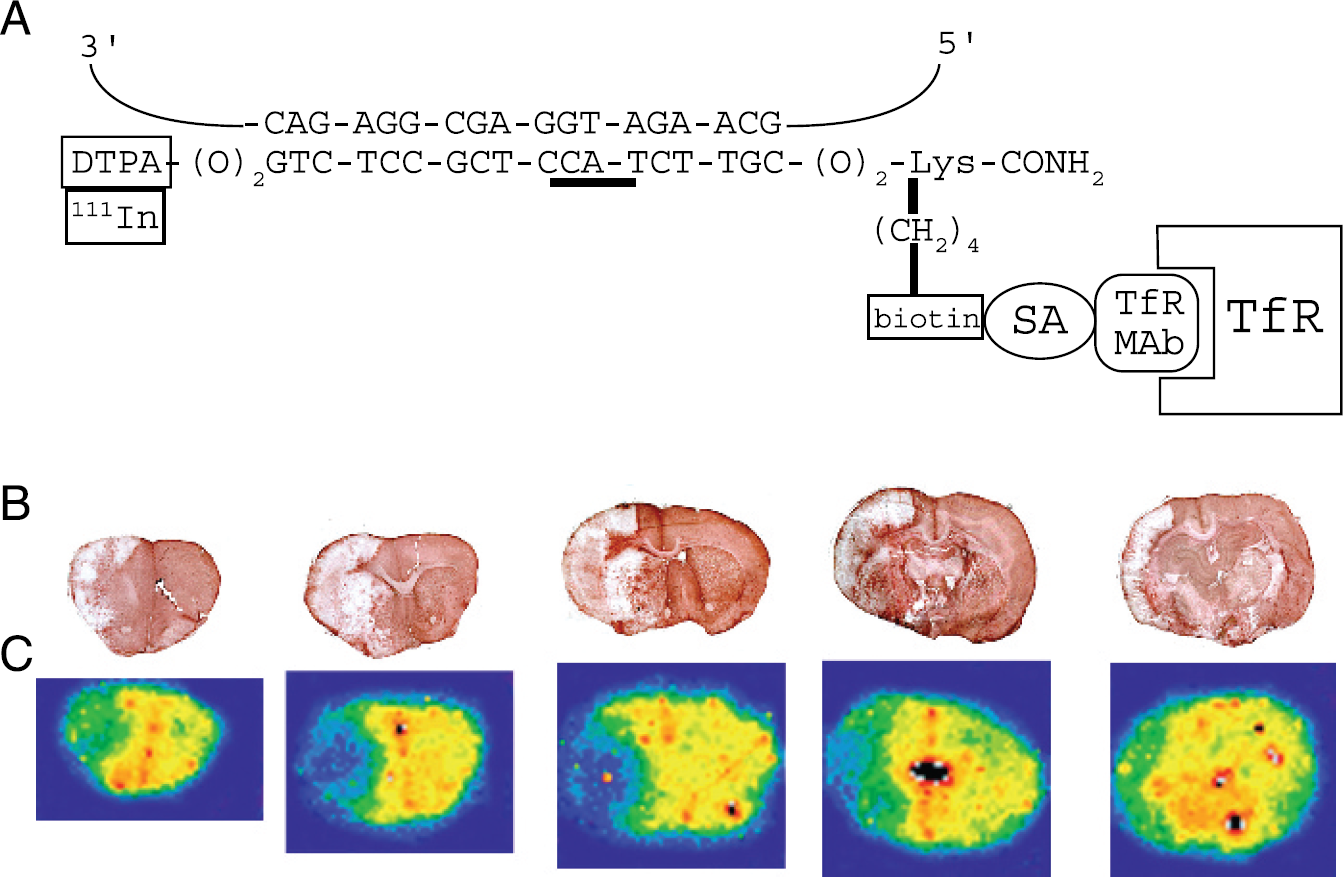
(A) Structure of [111In]-GFAP–PNA/SA–OX26 is shown, and is comprised of an 18-mer PNA antisense to nucleotides 20–37 of the rat GFAP mRNA (accession number NM_017009); the sequence antisense to the ATG methionine initiation codon of the GFAP mRNA is underlined. The amino terminus of the PNA is composed of two linkers and a DTPA moiety, which chelates the 111In; the nine-atom linker (–O–) is –CH2–O–(CH2)2–O–(CH2)2–O–CH2–. The carboxyl terminus is composed of two linkers, a lysine (Lys) residue, and an amidated carboxyl terminus; the e-amino group of the Lys is biotinylated, which enables conjugation to SA. The SA is conjugated to the OX26 anti-rat TfR murine MAb via a stable thio-ether linkage. (B) Immunocytochemistry (ICC) with an anti-GFAP antibody of serial coronal sections of rat brain examined at 23 hr following a 1-hr MCAO. The infarcted region shows decreased expression of immunoreactive GFAP. (C) Film autoradiograms of the coronal sections taken from the same blocks used for the ICC in panel B. Rats were subjected to 1 hr of MCAO followed by 23 hr of reperfusion. The animals were then reanesthetized and injected intravenously with [111In]-GFAP–PNA/SA–OX26, and euthanized 1 hr after isotope injection. Brains were frozen for film autoradiography.
Targeted PNA antisense radiopharmaceuticals can be radiolabeled with 111-Indium if the PNA is conjugated with a chelator moiety such as diethylenetriaminepentaacetic acid (DTPA) to enable radiolabeling with 111-Indium (Figure 1A). A [111In]-targeted PNA antisense radiopharmaceutical was used to image the up-regulation of gene expression in experimental brain cancer [13]. The purpose of the present studies was to test the hypothesis that targeted PNA antisense radiopharmaceuticals labeled with 111-Indium could also be used to image the down-regulation of target gene expression. One model of down-regulation of gene expression in brain is experimental regional brain ischemia, which is associated with decreased expression of the gene encoding glial fibrillary acidic protein (GFAP) at 24 hr following the ischemic insult [14]. For the present studies, an 18-mer PNA was designed that was antisense to nucleotides 20–37 of the rat GFAP mRNA, which is the region that surrounds the methionine initiation codon. The carboxyl terminal lysine residue of the PNA was monobiotinylated, which allowed for high affinity attachment of the PNA to a conjugate of streptavidin (SA) and the OX26 MAb to the rat TfR (Figure 1A).
The target GFAP mRNA is produced in the cytosol of brain cells, and there are two principal membrane barriers separating the circulating PNA from the cytosolic compartment of brain cells [10]. The first membrane barrier that must be traversed is the brain capillary endothelial plasma membrane, which forms the BBB in vivo. Owing to high expression of TfR on the brain capillary endothelial plasma membrane [15], the OX26 MAb to the rat TfR undergoes receptor-mediated transcytosis across the BBB in vivo [16,17]. The TfR is also expressed on neurons and astrocytes in the brain [18,19], and the TfR MAb binds the brain cell TfR to trigger receptor-mediated endocytosis of the PNA–MAb complex into the cytosolic compartment of brain cells [9]. Within the cytosol, the radiolabeled PNA may then hybridize via a sequence-specific mechanism to the target mRNA. This hybridization causes a selective sequestration of the radiopharmaceutical in the region of interest. Because the GFAP transcript is widely expressed in astrocytes throughout the brain, there should be a generalized retention of the anti-GFAP PNA radiopharmaceutical in the brain that expresses normal levels of the GFAP transcript. However, the GFAP gene is not expressed in the core of a cerebral ischemic infarct in the first 24 hr after a reversible middle cerebral artery occlusion (MCAO) [14]. The present studies examine whether the down-regulation of GFAP gene expression in the MCAO model can be detected with a targeted PNA [111In]-radiopharmaceutical.
Materials and Methods
Materials
111In-Cl3 (74 MBq [2 mCi]) was purchased from Perkin Elmer Life and Analytical Sciences (Boston, MA). The PNA was custom synthesized by Applied Biosystems (Bedford, MA), and the molecular weight of the PNA was confirmed by mass spectrometry. The PNA was antisense to the rat GFAP mRNA, and was designated GFAP–PNA. The GFAP–PNA contained a DTPA moiety at the amino terminus, and a biotin group on the ε-amino group of the lysine residue at the carboxyl terminus. Following labeling with 111-Indium, this PNA was designated [111In]-GFAP–PNA. A Superose 12 HR 10/30 fast protein liquid chromatography (FPLC) column was obtained from Pharmacia Biotech (Piscataway, NJ). Chelex-100 resin was obtained from Bio-Rad Laboratories (Hercules, CA). The rabbit anti-Glut1 glucose transporter polyclonal antiserum was prepared against a synthetic peptide encoding the 13 amino acids at the carboxy terminus of the rat or human Glut1 isoform as described previously [20], and this antiserum was designated rabbit anti-Glut1. The OX26 MAb was purified from hybridoma-generated ascites with protein G affinity chromatography. Secondary antibodies Alexa Fluor 488 donkey anti-mouse IgG (fluorescein channel) and Alexa Fluor 594 donkey anti-rabbit IgG (rhodamine channel) were purchased from Molecular Probes (Eugene, OR, USA). Vectashield and biotinylated horse anti-mouse IgG preabsorbed with rat IgG were from Vector Labs (Burlingame, CA, USA). Recombinant streptavidin (SA), the murine anti-GFAP MAb (G-3893), and all other reagents were obtained from Sigma (St. Louis, MO).
Conjugation of Streptavidin to the OX26 MAb
The tetrameric recombinant SA was conjugated to the murine OX26 MAb to the rat TfR with a stable thio-ether linkage, and purified with a Sephacryl S300 gel filtration column as described previously [8]. This conjugate is designated SA–OX26 and is a 1:1 conjugate of SA and the OX26 MAb, and has 3.3 biotin binding sites per SA–OX26 conjugate. There is immediate capture of a biotinylated PNA by the SA–OX26 conjugate. Following mixing of SA–OX26 with [111In]-GFAP–PNA, the final conjugate is designated [111In]-GFAP–PNA/SA–OX26.
Radiolabeling of GFAP-PNA with [111In]
Trace heavy metals were removed from water used to prepare all buffers by pretreatment with Chelex-100. The 111In (55.5 MBq, 1.5 mCi, 34 pmol) was added to the GFAP–PNA (328 pmol) in a total volume 1.5 mL of Chelex-treated PBST (0.01 M Na2HPO4, 0.15 M NaCl, pH = 7.4, 0.05% Tween-20). After 30 min at room temperature, the SA–OX26 was added to the [111In]-GFAP–PNA. Radiolabeling of the PNA with [111In] was confirmed by Sephadex G-25 size-exclusion chromatography in Chelex-treated PBST. The conjugate of [111In]-PNA and SA–OX26 was prepared by mixing the [111In]-PNA (328 pmol) with 100 μg of SA–OX26 (500 pmol) for 30 min at room temperature. Binding of the [111In]-PNA to the SA–OX26 was confirmed by a gel filtration FPLC with a single Superose 12 HR 10/30 column and isocratic elution in PBST at a flow rate of 0.25 mL/min.
Reversible Middle Cerebral Artery Occlusion
All animal protocols were approved by the UCLA Animal Research Committee. Adult male Sprague–Dawley rats (body weight, 310–350 g) were purchased from Harlan Breeders (Indianapolis, IN). Focal cerebral ischemia was produced by intraluminal MCAO, as described previously [21]. The rats were anesthetized by intraperitoneal injection of chloral hydrate (400 mg/kg), which was repeated in 100 mg/kg doses as required throughout the surgery. The body temperature was maintained in the normal range (37°C) with a Harvard thermal blanket with a rectal probe (Harvard Apparatus, Holliston, MA) during surgery, and hypothermia was not observed. The right common carotid artery and the right external carotid artery were exposed and the occipital artery and superior thyroid artery were electro-coagulated. The right pterygopalatine artery was ligated, and the right common carotid artery was clamped, and a 4–0 nylon suture was inserted retrogradely via arteriectomy of the external carotid artery into the internal carotid artery. Before insertion, the tip of the 5-cm nylon suture was rounded by heating near a flame and was coated with 0.1% poly-l-lysine solution and dried in a 60°C oven for 1 hr [22]. The suture was slowly advanced until resistance was felt. After the intraluminal suture was inserted, the external carotid artery was ligated and common carotid artery clamp was released. The neck skin incision was sutured, leaving 10 mm of nylon silk protruding. The animal was kept warm with a heating lamp. One hour after ischemia, the intraluminal nylon suture was carefully withdrawn to allow for reperfusion. Following surgery, the animals were kept warm with a heating lamp for further recovery and returned to their cages. The animals were injected with targeted antisense radiopharmaceutical at 23 hr following the 1-hr reversible MCAO.
Film Autoradiography
At 23 hr after the 1-hr period of ischemia, rats were reanesthetized with chloral hydrate for the intravenous injection of brain imaging agent. The rats were intravenously injected with 5.5 MBq (150 μCi) of [111In]-GFAP–PNA/SA–OX26 via the femoral vein. The animals were decapitated at 1 hr after injection, and the brain was rapidly removed from the cranium, chilled in a freezer for 20 min, and 6 × 2-mm thick coronal slices were prepared with a brain matrix (ASI Instruments, Warren, MI), frozen in powdered dry ice, dipped in Tissue-Tek optimal cutting temperature (OCT) embedding compound (Sakura Finetek, Torrance, CA), and blocks were stored at −70°C. Frozen sections of 20-μm thickness were prepared on a Mikrome HM 505E cryostat (Mikron Instruments, San Diego, CA), mounted on glass slides, and dried at room temperature. For film autoradiography, the brain sections were exposed with intensifying screen to Biomax MS film (Kodak, Rochester, NY) at −70°C for 7 days. The films were then developed for 1–2 min using Kodak developer and fixed for 5 min using Kodak fixer. The film was scanned in a 1200 dpi PowerLookIII scanner (Umax Data Systems, Hsinchu, Taiwan) with transparency adapter and cropped in Photoshop 5.5 on a G4 Power Macintosh. The integrated density over either the infarct region or the contralateral brain was quantified using NIH Image 1.62 software, and normalized by pixel area of the scanned region.
Cerebral Blood Flow
At 23 hr after the 1-hr MCAO, the rat was anesthetized again with chloral hydrate (400 mg/kg) intraperitoneally. The left femoral artery was cannulated with heparinized PE-50 tubing, which was connected to a 3-mL heparinized syringe. The syringe was attached to a withdrawal pump (Harvard Apparatus, Dover, MA). The right femoral vein was catheterized with a second heparinized PE-50 tubing and injected with 0.2 mL of Ringers–Hepes buffer/pH 7.4 containing 0.1% rat serum albumin and 5 μCi of 3H-diazepam (86.0 Ci/mmol, Perkin Elmer). Simultaneous with the rapid (<0.5 sec) intravenous injection, the syringe pump was turned on at a rate of 1.0 mL/min for arterial blood withdrawal. Immediately at 30 sec after the injection, the animal was decapitated and the pump was turned off. The rat brain was quickly removed, briefly chilled, and 6 × 2 mm coronal slices were prepared with the rat brain matrix. The brain sections were stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC) at 37°C for 15 min. The TTC stained viable brain tissue dark red, whereas infarcted tissue was unstained. The infarcted tissue from each of the six sections was dissected with forceps, pooled and weighed. A comparable amount of noninfarcted tissue was also dissected from the contralateral brain of each section, pooled and weighed. The brain tissue and aliquots of whole blood were solubilized with Soluene 350 (Perkin Elmer), and neutralized with glacial acetic acid prior to liquid scintillation counting. The cerebral blood flow (CBF) was computed by dividing the dpm/g brain by the dpm/μL whole blood and the time period (0.5 min), and reported as μL/min/g.
Immunocytochemistry and Confocal Microscopy
Confocal microscopy with the OX26 anti-TfR and anti-Glut1 antibodies was performed on brain sections that were fixed at −20°C with 100% acetone for 20 min and air dried for 120 min followed by washing in PBST. Slides were blocked with 10% donkey serum in PBS for 60 min at room temperature. Primary antibodies were added in the following concentration diluted in 0.01M PBS containing 3% bovine serum albumin: OX26 (10 μg/mL) and anti-Glut-1 (1:1000). Control sections consisted of rabbit preimmune serum (1:1000) and mouse-IgG2a (10 μg/mL). Sections were incubated overnight at 4°C in a humidified chamber and washed with PBS after warming to room temperature. Secondary antibodies added were Alexa Fluor 488-conjugated donkey anti-mouse IgG and Alexa Fluor 594-conjugated donkey anti-rabbit IgG, both in a concentration of 5 μg/mL. After 60 min, slides were rinsed with PBS thoroughly and cover-slipped with Vectashield. Confocal imaging was performed employing a Zeiss LSM 5 PASCAL confocal microscope with dual argon and helium/neon lasers equipped with Zeiss LSM software for image reconstruction. All sections were scanned in multitrack mode to avoid overlap of the fluorescein (excitation at 488 nm) and rhodamine (excitation at 568 nm) channels. Pinhole size for each channel was kept as small as possible as to ensure sufficient signal-to-noise ratio and highest spatial resolution. Detector gain and amplifier offset were optimized to reduce artificial background for each image. No amplifier gain was used. All image slices were scanned with a 1024 × 1024 resolution. For acquisition of three-dimensional images, six serial images with a slice thickness between 1.6 and 3.7 μm were used. For all control studies, all scanning parameters were kept constant.
Immunocytochemistry with the anti-GFAP antibody was performed on brain sections fixed with 4% paraformaldehyde at 4°C for 20 min. Sections were treated with the anti-GFAP primary antibody (1 μg/mL) and the secondary biotinylated horse anti-mouse antibody (10 μg/mL) as described previously [23], and were not counterstained. Prior to cover slipping, the dried immunostained sections were scanned in parallel with scanning of the X-ray films described above.
Results
The down-regulation of GFAP gene expression within the infarct core was examined by GFAP immunocytochemistry. The brain was removed at 23 hr following a 1-hr MCAO and divided into five coronal sections, which were immunostained with a GFAP antibody (Figure 1B). Although the immunoreactive GFAP was detected in a noninfarcted brain, GFAP expression was decreased in the region of the infarct (Figure 1B).
Adult rats were subjected to a 1-hr MCAO followed by 23 hr of reperfusion, and were then administered intravenously with the [111In]-GFAP–PNA/SA–OX26 radiopharmaceutical. The animals were euthanized 60 min following administration of the radiopharmaceutical, and the sequestration of regional brain radioactivity in coronal brain sections was detected by film autoradiography (Figure 1C). The diminished in vivo retention of radioactivity in the brain in the region of the infarction (Figure 1C) paralleled the down-regulation of GFAP expression observed in autopsy brain sections (Figure 1B). The relative integrated density per square area over the infarct region and the contralateral brain was 1.0 ± 0.1 and 3.9 ± 0.3, respectively.
The decreased uptake of the targeted PNA radiopharmaceutical could be due to a generalized down-regulation of microvascular TfR in the region of the infarct. Therefore, brain sections were examined with confocal microscopy using antibodies to both the TfR and to an endothelial-specific marker, the Glut1 glucose transporter. The capillaries within the core of the infarct express high levels of immunoreactive TfR as shown at both low magnification (Figure 2A) and high magnification (Figure 2D). The capillaries within the infarct core also express the endothelial Glut1 glucose transporter (Figure 2B and E). The overlap image showed co-localization of the BBB TfR and Glut1 glucose transporter (Figure 2C and F). The immune staining in the contralateral brain for the TfR and Glut1 is shown in Figure 2G and H, respectively, and the overlap image is shown in Figure 2I. There was no immunolabeling of the brain with the preimmune antibodies corresponding to either the TfR antibody or the Glut1 glucose transporter antibody.
The decreased uptake of the targeted PNA could be due to reduced CBF in the infarct region at 23 hr after the 1-hr MCAO. Therefore, rats were subjected to a 1-hr MCAO, and 23 hr after the restoration of brain blood flow, the animals were reanesthetized and administered [3H]-diazepam, a fluid microsphere. CBF was measured with the external organ technique (Materials and Methods). The brain sections were stained with TTC to identify the infarct region and the contralateral brain, as shown in Figure 3A. There was no significant change in CBF in the infarct region versus the contralateral brain at 23 hr after the restoration of brain blood flow (Figure 3B).
Discussion
The results of the present studies are consistent with the following conclusions. First, the decrease in GFAP gene expression that occurs 24 hr following reversible brain ischemia can be detected in vivo with imaging of gene expression using an antisense radiopharmaceutical, provided membrane targeting technology is used (Figure 1C). Second, the capillaries perfusing the ischemic core 24 hr after the reversible ischemia continue to express high levels of brain endothelial markers such as the TfR or the Glut1 glucose transporter (Figure 2).
The [111In]-GFAP–PNA is not taken up by brain following an intravenous injection, owing to lack of transport of the PNA across the BBB [13]. However, if the GFAP–PNA is conjugated to a targeting vector such as the TfR MAb, then there is generalized sequestration of radioactivity within the brain in vivo, as the TfR MAb mediates uptake of the PNA antisense radiopharmaceutical across both the BBB and the brain cell membrane [13]. The uptake of the [111In]-GFAP–PNA/SA–OX26 in the noninfarcted region of brain is demonstrated in the present study, as shown by the film autoradiography of brain removed 60 min after intravenous injection of the targeted radiopharmaceutical (Figure 1C).
The selective retention of the PNA antisense agent in the brain requires the concerted action of three processes: (i) influx from blood to brain of the targeted antisense radiopharmaceutical, (ii) efflux from brain back to blood of the antisense radiopharmaceutical, and (iii) selective retention of the antisense radiopharmaceutical in regions of brain that express the target mRNA [10]. Conjugation of the PNA to the TfR MAb enables the influx of the PNA from blood across both the BBB and the brain cell membrane, because both membranes express the TfR. The TfR also mediates the efflux of the MAb from brain back to blood [24]. In addition, the BBB expresses an Fc receptor on the brain side of the BBB that causes the rapid efflux of IgG molecules from brain back to blood [25]. The ability of the targeted PNA radiopharmaceutical to both influx and efflux across the BBB allows for selective retention of the PNA in the region of the brain expressing the target mRNA [10]. However, it must be shown that the PNA still hybridizes to the target mRNA despite conjugation to the SA–MAb targeting system. Prior work produced cloned GFAP RNA by in vitro transcription and the hybridization of the [111In]-GFAP–PNA/SA–OX26 to the GFAP RNA was examined by Northern blotting [13]. The Northern blots showed the GFAP–PNA still hybridizes to the target GFAP mRNA despite conjugation to the SA–OX26 delivery system. Binding of the biotinylated PNA to the SA–OX26 causes no steric interference and no inhibition of GFAP–PNA hybridization to the target mRNA [13].

Confocal microscopy of the cortical infarct (A–F) and the contralateral brain (G–I) at 23 hr after a 1-hr reversible MCAO is shown for the TfR and the Glut1 glucose transporter. Immune staining is shown for the TfR (green) in panels A and D for the infarct and in panel G for the contralateral brain. Immune staining is shown for the Glut1 glucose transporter (red) in panels B and E for the infarct and in panel H for the contralateral brain. The TfR/Glut1 overlap images are shown in panels C and F for the infarct and in panel I for the contralateral brain. Immune staining of brain following incubation with a nonimmune mouse isotype IgG2a and rabbit IgG was also performed, and there was no immune staining of brain with the control antibodies. Images A–C and G–I were taken with a 20× objective, and the magnification bar is 20 μm. Images D–F are 3-D projection views of six consecutive planar images.
The [111In]-GFAP–PNA/SA–OX26 was sequestered in regions of brain outside the infarct zone following intravenous administration (Figure 1C), owing to generalized expression of GFAP mRNA in the brain outside the infarct (Figure 1B). However, in the region of the cerebral infarction caused by a 1-hr occlusion of the middle cerebral artery, there is down-regulation of radioactivity in the region of brain that overlaps with the area of decreased GFAP gene expression shown by immunocytochemistry (Figure 1B and C). Quantitation of autoradiogram signal showed the brain radioactivity in the contralateral brain was 390% of the radioactivity in the infarct zone (see Results). These studies provide an in vivo corroboration of prior work on autopsy brain showing that a 1-hr reversible brain ischemia is associated with a decrease in expression of both the immunoreactive GFAP protein [14] and the GFAP mRNA at 24 hr after the ischemic insult [26]. In contrast, at 7 days following the ischemic insult, when the infarct region is infiltrated with an astrogliotic reaction, there is an increase in astrocytic GFAP gene expression [14]. The decreased uptake of the targeted GFAP–PNA radiopharmaceutical in the region of the infarct is not due to a down-regulation of the brain microvascular endothelial TfR in the core of the infarct. High expression of the microvascular TfR, in parallel with an endothelial marker, the Glut1 glucose transporter, in the ischemic infarct core is demonstrated by confocal microscopy (Figure 2). In this model, the middle cerebral artery is occluded for 60 min followed by 23 hr of reflow through the middle cerebral artery. The findings of persistent expression of both the TfR and the Glut1 glucose transporter in capillaries perfusing the ischemic core are consistent with other results showing that rates of CBF are normalized within 90 min after a reversible occlusion of the middle cerebral artery [27]. The normalization of CBF in the reversible MCAO model is confirmed by the direct measurements of CBF using [3H]-diazepam in both the infarct and the contralateral brain (Figure 3).

(A) TTC stains of six parallel consecutive coronal sections of rat brain removed 23 hr after a 1-hr MCAO. The TTC dye stains healthy tissue red, which appears black in the grayscale image, and leaves infarcted tissue unstained. The most rostral section is on the left side, and the most caudal section of brain is on the right side. The TTC staining was used to separate the infarcted brain from the contralateral brain for measurements of CBF with [3H]-diazepam. (B) CBF data are mean ± SE (n = 4 rats).
In summary, these studies demonstrate it is possible to image the down-regulation of target gene expression in the brain using the MCAO model of regional brain ischemia, provided the PNA antisense radiopharmaceutical is attached to a trans-membrane targeting system (Figure 1A). The antisense PNA radiopharmaceutical is too polar to cross either the BBB or the brain cell plasma membrane, and the intravenous administration of a nontargeted PNA antisense radiopharmaceutical results in no detectable radioactivity in brain [8–10,13]. However, if the PNA antisense radiopharmaceutical is reformulated with drug targeting technology, then gene expression in the brain can be imaged in vivo. The present studies showing decreased GFAP gene expression in experimental stroke and prior work showing increased caveolin-1α gene expression in experimental brain cancer [13] provide evidence that either down- or up-regulation of gene expression in the brain can be imaged in vivo with targeted antisense radiopharmaceuticals labeled with 111-Indium.
Footnotes
Acknowledgments
This work was supported by a grant from the U.S. Department of Energy and by NCI grant R21-CA91098. The present address of T. S. is College of Pharmacy, Nihon University, Chiba, Japan. The present address of F. S. is Department of Neurology, Regensburg University, Regensburg, Germany. F. S. was supported by a grant from the Regensburger Forschungsförderung in der Medizin (ReForM, Regensburg, Germany).