
Abstract
We report the development of a novel dual-modality fusion reporter gene system consisting of Escherichia coli xanthine phosphoribosyltransferase (XPRT) for nuclear imaging with radiolabeled xanthine and Discosoma red fluorescent protein for optical fluorescent imaging applications. The dsRed/XPRT fusion gene was successfully created and stably transduced into RG2 glioma cells, and both reporters were shown to be functional. The level of dsRed fluorescence directly correlated with XPRT enzymatic activity as measured by ribophosphorylation of [14C]-xanthine was in vitro (Ki = 0.124 ± 0.008 vs. 0.00031 ± 0.00005 mL/min/g in parental cell line), and [*]-xanthine octanol/water partition coefficient was 0.20 at pH = 7.4 (logP = 0.69), meeting requirements for the blood-brain barrier (BBB) penetrating tracer. In the in vivo experiment, the concentration of [* C]-xanthine in the normal brain varied from 0.20 to 0.16 + 0.05% dose/g under 0.87 + 0.24% dose/g plasma radiotracer concentration. The accumulation in vivo in the transfected flank tumor was to 2.4 ± 0.3% dose/g, compared to 0.78 ± 0.02% dose/g and 0.64 ± 0.05% dose/g in the control flank tumors and intact muscle, respectively. [14C]-Xanthine appeared to be capable of specific accumulation in the transfected infiltrative brain tumor (RG2-dsRed/XPRT), which corresponded to the 585 nm fluorescent signal obtained from the adjacent cryosections. The images of endogenous gene expression with the “sensory system” have to be normalized for the transfection efficiency based on the “beacon system” image data. Such an approach requires two different “reporter genes” and two different “reporter substrates.” Therefore, the novel dsRed/XPRT fusion gene can be used as a multimodality reporter system in the biological applications requiring two independent reporter genes, including the cells located behind the BBB.
Introduction
Almost a decade has passed since the first report describing the paradigm and proof-of-principle for noninvasive reporter gene imaging [1]. Molecular-genetic imaging has grown rapidly and is complementing other investigative methods of modern biology; it is currently being applied to studies of gene expression regulation [2], activity of signal transduction pathways [3], angiogenesis [4], tumor metastasis [5,6], stem cell migration [7], and monitoring cells involved in different components of immune response [8].
Despite the broad spectrum of biological applications of reporter gene imaging, existing reporter gene-reporter substrate systems are limited with respect to studies of the central nervous system (CNS). This is largely due to the presence of the blood-brain barrier (BBB), which limits the passage and access of non-carrier-mediated hydrophilic probes to the reporter gene product. The basic requirements of a reporter gene-reporter probe imaging system were described in our original publication [1]. An additional requirement for the brain is that the reporter probe (tracer) should be sufficiently lipophylic to penetrate the BBB by passive diffusion, or be a substrate for a transporter in the brain microvascular endothelium (e.g., GLUTI for FDG) [9]–[11]. Many pathological processes in the CNS cause breakdown of the BBB (e.g., brain tumors), which provides the physiologic means for the contrast-enhanced imaging (e.g., T1 MRI with Gd-DTPA). However, many neuropsychiatrie diseases [12] and even some brain tumors are not always associated with BBB disruption. For example, low-grade gliomas show little BBB damage [13], and glioblastoma multiforme (GBM) is a very heterogenous brain neoplasm; often, some portions of GBMs do not exhibit BBB disruption and are not contrast enhancing, especially the infiltrative portions [14,15].

The concept of dsRed/XPRT reporter gene imaging. Radiolabeled [*]-xanthine freely crosses cell membrane and rapidly equilibrates between extra- and intracellular space. In the cytozole, xanthine is phosphoribosylated by the RedXPRT fusion protein also using phospho-ribosylpyrophosphate (PRPP). The product of the reaction, XMP, is irreversibly trapped within the cell, resulting in the accumulation of the radiolabel necessary for nuclear imaging. The DsRed domain of the reporter protein provides optical imaging.
Currently, there are no effective reporter gene-reporter probe imaging systems that satisfy all requirements for optimal imaging in the CNS. The only reporter gene imaging system that satisfies several, but not all of these requirements, is the human dopamine 2 receptor (hD2R)–[18F]-FESP system [16,17]. Although [18F]-FESP does penetrate the BBB and is selectively accumulated in hD2R-expressing cells, it is avidly accumulated by the dopaminergic neurons in the caudate nucleus, and this could significantly impact on the specificity of reporter imaging in the brain with [18F]-FESP.
Our objective was to develop a novel reporter gene-reporter probe system for imaging different molecular-genetic processes in the CNS using nuclear (PET) and optical (microscopy, FACS, OFI) imaging techniques. Specifically, we sought to develop a system in which the reporter probe would have a sufficient passive BBB permeability, rapid clearance from the circulation, and options for labeling with 18F or 11C.
We selected the Escherichia coli xanthine phosphoribosyltransferase (XPRT) and radiolabeled xanthine, because XPRT effectively ribophosphorylates xanthine. Xanthosine monophosphate (XMP), a nucleotide, is polar, and it is trapped inside the cell similar to FDG-6-phosphate (hexokinase-FDG metabolic imaging) and FIAU-monophosphate (HSV1-tk-FIAU reporter system). Xanthine is a suitable brain tissue imaging probe, because it belongs to the xanthanoid family of compounds, which includes caffeine and theophylline, and readily crosses the BBB having high volumes of distribution in the brain tissue due to their high logP values (−0.0403 and −0.0335, respectively) [18]–[20]. Xanthine is not a substrate for mammalian enzymes, except xanthine oxidase (XO), a ubiquitously expressed enzyme that catalyzes rapid oxidation of xanthine to uric acid, which is cleared via urinary excretion [21]–[23]. Furthermore, the metabolism of xanthine and the formation of uric acid can be effectively blocked by allopurinol-adrug that inhibits XO and is widely used for the treatment of gout [21]–[23].
Studies involving a native, intact BBB could not be performed because no transgenic animals expressing XPRT or RedXPRT currently exist. Therefore, we used a RedXPRT-transduced intracranial glioma model to assess the accumulation of [14C]-xanthine in transduced tumors. The RG2 intracerebral glioma model was selected, because RG2 gliomas have relatively low BBB disruption (low passive permeability) and no Gd-DTPA contrast enhancement of Tl-weighted MRI at the early stages of the infiltrative intracerebral growth of this tumor cell line [9,24,25].
In the current study, we developed the XPRT and DsRed fluorescent protein fusion gene (RedXPRT) and assessed the sensitivity and specificity of this new reporter gene using [14C]-xanthine, [14C]-8-bromo-xanthine, and [3H]-hypoxanthine in RedXPRT-transduced RG2 glioma cells (RG2/RedXPRT) in vitro (Figure 1). We also generated and tested a mutant variant of XPRT in fusion with DsRed, where the carboxy-terminus amino acid sequence containing the lyzosomal-localizing signal of XPRT [26] was truncated in an attempt to improve the metabolic availability of the reporter protein. We demonstrated that the RedXPRT-radiolabeled xanthine reporter gene-reporter probe approach was effective, sufficiently sensitive, and suitable for in vivo imaging of different molecular-genetic processes in the CNS. Herewith, we discuss the potential routs of synthesis of [11C], [76Br], and [18F] derivatives of xanthine, which could broaden the spectrum of applications of this new reporter gene system.
Materials and Methods
Generation of dsRed/XPRT Reporter Gene
All DNA manipulations were performed according to standard procedures [27] using restriction enzymes, T4 DNA ligase, CIP, and buffers according to manufacturers' instructions (Life Technologies, Roche, and New England BioLabs, Beverly, MA).
First, E. coli XGPRT cDNA was amplified from the plasmid pMVGT (ATCC, Manassas, VA) using primers 5′-AGTCAAGCTTATGAGCGAAAAATAC-3′ and 5′-AGTCATCGATTTAGCGACCGGAGATTG-3′ and was subcloned into pLNCX retroviral vector (Clontech, CA) between HindIII and ClaI restriction sites downstream of internal pCMV promoter. To receive the fusion (Red1-XGPRT) gene with DsRed1 fluorescence protein, we obtained the fragment from the pDsRed1 plasmid with SnaBI and XhoI enzymes and ligated this fragment with plasmid pLNCX-XGPRT digested by SnaBI and HindIII and a small 32-bp DNA fragments coding of eight Gly junctions between two genes and an incorporated site for restriction NotI (5′-TCGAGGAGGGGGTGGCGGAGGTGGGCGGCCGCA3′) with XhoI and HindIII sites for ligation. After three pieces of ligation and minipreps analysis, we obtained a new plasmid pLNCXRed1-8G-XGPRT, which we used for further work (Figure 2).
To get a mutant form of XGPRT with truncated 9 and 14 amino acids C-terminus, sense oligonucleotide, 5′-AGGTGGCGGCCGCAAGCTTATGAGC-3′, and an antisense primers, 5′-ATGCGTGAATCGATTTAGACGCCCATATCCCACG-3′ (−9 Cla) and 5′-ATGCGTGATCGATTTACGGCTGTTCAATCCAG-3¶ime; (−14 Cla) containing restriction sites for Notl and ClaI (italicized), respectively, were used to amplify a 434-bp fragment for the mutant form of gene without 14 C-terminal amino acids and a 449-bp fragment for the mutant form of gene without 9 amino acids on the C-terminus by PCR (Figure 2B). After digestion of PCR fragments by corresponding restriction enzymes, the fragments were ligated with pLNCXRed1-8G-XPRT plasmid restricted by NotI and ClaI after which the wild type of gene XGPRT was replaced on the mutant forms. The fidelity of the RedI-XPRT and the mutant forms sequence generated by PCR was verified by DNA sequencing.
Transfection and Selection of the Cell Lines
RG2 rat glioblastoma cell line was kindly provided by Dr. D. Bigner (Duke University, Durham, NC) and was transfected using retroviral supernatant collected form SVG-pseudotyped retroviral producer cell line in the presence of polybrene 8 μg/mL [28]. A mixed population of transduced RG2/RedXPRT-positive cells was obtained by selection with G418 at a dose of 1000 mg/1 with subsequent cultivating under selective pressure of 500 mg/1 of Geneticin (Life Technologies, Rockville, MD).
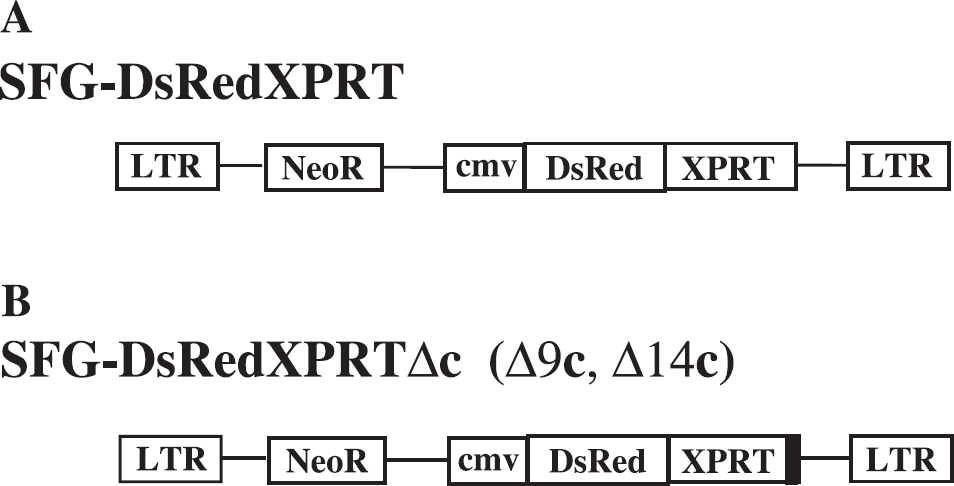
Vector design. (A) The MoMLV retroviral vector backbone bearing RedXPRT fusion reporter gene under CMV promoter for constituitive expression. (B) The MoMLV retroviral vector containing C-terminal 9 or 14 amino acids truncated forms of XPRT gene in fusion with DsRed.
In Vitro Assessment of RedXPRT Reporter Gene Expression
In vitro assessment of the XPRT gene expression was performed using a radiotracer accumulation assay as previously described [1] with [8-3H]-xanthine (9.9 Ci/mmol) or [8-14C]-xanthine (55 mCi/mmol). [8-14C]-Hypoxanthine (56 mCi/mmol) or [2,8-3H]-hypoxanthine (24.5 Ci/mmol), respectively, were used as a second radiotracer in dual-label experiments to normalize the xanthine uptake data to the level of activity of the endogenous hypoxanthine phosphoribosyltransferase (which has no affinity to xanthine [21]–[23]). All radiotracers were obtained commercially (Moravek Biochemicals, Brea, CA).
To assess the potential tracer-catabolizing effects of XO in cells, the dual-label in vitro uptake experiments were performed after preincubation of the cell cultures for 2 hr with allopurinol 100 μg/mL—a well-known XO inhibitor [21]–[23]. [14C]-Bromoxanthine (56 mCi/mmol) was custom-synthesized by Moravek Biochemicals and studied in the same settings, as an alternative candidate for imaging tracer probe.
Additional in vitro assessment of RedXPRT expression was performed using fluorescence microscopy and FACS of the dsRed subunit in the same cell cultures in which the radiotracer uptake had been performed. Fluorescence microscopy studies were performed in live cultures growing in tissue culture flasks (Costar, Corning, NY) on a Nikon Eclipse TS 100 inverted fluorescence microscope equipped with an Orca II CCD (Hamamatsu, Japan) linked to a digital image analysis system—MCID 5.5 (Imaging Research, Ontario, Canada).
For the quantitative flow cytometry studies, cells were harvested and analyzed using FACSCalibur (Becton Dickinson, NJ), using 535 nm excitation and 585 nm emission light (phycoerythrine channel). The settings of FACS analysis were preserved throughout the study with different clones of dsRed/XPRT-expressing cells. The mean fluorescence was correlated with the rate (Kin value) of the [14C]XPRT accumulation in the same cell cultures.
Measurements of Xanthine Lipophilicity
To measure the lipophilicity of xanthine, the octanol/water coefficient was determined and logPoctanol was calculated. [14C]-Xanthine (Moravek Biochemicals; 56 mCi/mmol, 97% pure) was added to a premixed emulsion containing equal volumes of octanol and water (pH = 7.4) at room temperature. The suspension was thoroughly vortexed and centrifuged at 12,000 × g for 5 min. Aliquots of the water and octanol fractions were obtained and counted in liquid scintillation counter (Tri-Carb, Perkin-Elmer, NJ). The octanol/water partition coefficient (PC) was calculated and expressed as a logPoctanol value for xanthine. The empirical value was compared to calculated logP (ClogP) values obtained through analyzing the chemical structure of xanthine and other xanthanoids (caffeine, theophylline, nicotine, uric acid, etc.) using software ChemDraw Ultra 2000 version 6.0.1 (Cambridge Soft, MA), and these values were used to estimate the permeability-surface area product of brain capillaries using the graphical relationships originally described by Davson and Danielli [29], and later modified by Fenstermacher and Rappoport [30]–[32]:

where the octanol/water PC is assumed to be a measure of lipid membrane solubility and Dm is the transmembrane and transcellular diffusivity, which is commonly approximated by the aqueous diffusion coefficient at 37°C or the reciprocal of the square root of the compound molecular weight (MW−1/2).
Assessment of BBB Permeability of Xanthine
To assess BBB permeability to xanthine in vivo, we compared cerebral blood flow (CBF) measurements obtained with xanthine and iodoantipyrine (TAP) in the same nu/nu athymic mice (Harlan, MA). IAP is an established tracer for measuring blood flow in rodents [33]–[35]. A solution containing 2.0 μCi of 4-iodo-N-methyl-[14C]antipyrine (IAP; specific activity: 40–60 mCi/mmol, American Radiolabeled Chemicals, St. Louis, MO) and 20 μCi of [3H] -xanthine (specific activity 3–10 mCi/mmol, Moravek Biochemicals) dissolved in 200 mL of 0.9% saline was injected intraperitoneally (n = 6 animals) [36]. Thirty seconds after bolus intraperitoneal injection of the tracer solution, experiments were terminated by decapitation, and the head of the animal was immediately immersed in liquid −80°C agent OOO (Fisher Scientific, NJ) to avoid diffusion of tracers (<15 sec). The terminal blood sample was processed for whole blood and plasma radioactivity, and the brain was dissected to obtain gray and white matter tissue for radioactivity measurements in a liquid scintillation counter (Tri-Carb 1600TR, Perkin-Elmer, MM) using an established dual isotope and external standard quench correction counting protocol.
CBF was calculated from the IAP data as described by Sakurada et al. [33] using the following equation:

where the ratio of local tissue radioactivity Ci(T) to local blood flow (Fi) equals the flow-related convolution integral of the time course of the arterial blood concentration [Cb], and the tissue/blood PC for IAP (λI) is 0.8 [33]. A ramp (linear) blood input function of IAP has been shown to exist over at least 30 sec following intraperitoneal injection of the tracer [36] (Myagawa et al., unpublished observations) and the input function profile could be approximated from the terminal blood sample. In contrast to IAP, xanthine is essentially bound or trapped within the brain during the 30-sec experiment and local CBF is calculated by the following equation [37]:

Subcutaneous and Intracerebral Tumor Models
To assess the radiographic and fluorescent detectability of subcutaneous RedXPRT-expressing tumors in vivo, we injected 106 RG2-dsRed/XPRT clone 4 cells in the dorsal aspect of the left thigh of nu/nu athymic mice (Harlan, Indianapolis, IN), as previously described [3]. The RG2/TKGFP cell line [38] was implanted into the contralateral thigh area, as a control.
To test the BBB permeability and the specificity of imaging XPRT by xanthine, we developed a dual intracerebral tumor model in which two intracerebral tumors were established in opposite hemispheres of the brain in the same animal: the RG2/RedXPRT, in the left hemisphere, and the RG2/TKGFP, in the right hemisphere. Using a short-beveled 30-ga needle attached to a Hamilton 50-μl syringe, tumor cells (105 cells in 10 μl of culture medium) were slowly injected into the brain parenchyma by stereotactic placement via the burr holes drilled 3 mm posterior and 2 mm lateral to bregma and to a depth of 4 mm below the dura mater. The surgical procedure was performed using a small animal stereotactic instrument (David Kopf Instruments, Tujunga, CA) under inhalation anesthesia (isoflurane 2% and oxygen 98%, 0.5 L/min).
MR Imaging of Intracerebral Tumor Model
To establish the appropriate tumor model and ensure the preservation of the BBB prior to the in vivo radiotracer study, the animals were evaluated by Tl-weighted contrast-enhanced MR imaging of the head, as previously described [39]. Briefly, the mice were anesthetized by intraperitoneal administration of ketamine (0.01 mg/kg) and acepromazine (0.05 mg/kg), then the mice were injected intravenously with Gd-DTPA (0.2 mL/kg) and imaged on the CSI 4.7T system (General Electric, PA) using standard echo sequences (TR = 300 msec, TR = 10 msec, four excitations per phase encoding step). Coronal slices (3 mm thick, 256 × 256 matrix) were obtained. Animals with areas of BBB disruption (contrast-enhancing regions on the T1-weighted brain images) were excluded from the current study.
In Vivo Fluorescence Assessment
To evaluate transcutaneous fluorescence imaging of RedXPRT-transduced subcutaneous tumors, 535 nm excitation filter equipped excitation light source (Lightools, Encinitas, CA) and CCD camera (Hamamatsu, Bridgewater, NJ) with 585 nm emission filter (Omega, Brattleboro, VT) were used, under general anesthesia (ketamine/acepromazine). The fluorescent images were digitally merged with regular black-and-white images obtained sequentially using MCID5 image analysis software (Imaging Research, Ontario, Canada).
Ex Vivo Assessment of dsRed/XPRT Expression Level
To assess dsRed/XPRT-radiolabeled xanthine reporter system in the animals bearing subcutaneous or intracerebral RG2/RedXPRT tumor xenografts, the mice were pretreated with allopurinol (300 μg/kg) for 12 hr and then injected intravenously with 30 mCi of [14C]-xanthine in 200 μl of isotonic solution. One hour after radiotracer administration, the mice were sacrificed by guillotine decapitation. The whole brain and each of the subcutaneous tumors with the underlying soft tissues were snap-frozen in liquid Freon (Fisher Scientific, Pittsburgh, PA) and embedded in the cryomatrix (Shandon Lipshaw, Pittsburgh, PA) for sectioning on a chryomicrotome (Hacker Instruments, Fairfield, NJ) to 20-μm-thick tissue sections.
Terminal blood and tissue samples were obtained, and radioactivity was measured using a β-spectrometer (Perkin-Elmer, CT). Quantitative autoradiography (QAR) was performed using [14C]-methylacrylate standards calibrated to 20-μm-thick sections, as previously described [1]. The autoradiograms were digitized using CCD camera (MTI Dage-725) and MCID5 digital image analysis system (Imaging Research). Every other cryo-section of the brain or subcutaneous tumor was fixed in ethanol/methanol (1:1) mixture for 30 sec, air-dried, and cover-slipped using the AquaMount medium (Fisher Scientific, PA). Fluorescent images of these tissue sections were obtained with an inverted fluorescence microscope (Eclipse TS 100, Nikon, Japan) equipped with an Orca II CCD (Hamamatsu, Japan) linked to a digital image analysis system—MCID 5.5 (Imaging Research). Coregistration of the QAR and fluorescence images and a comparative image analysis were performed using the MCID 5.5 system.

In vitro fluorescence of RG2 cell lines transfected with dsRed/XPRT C-terminal mutant forms. (A) Fluorescent microscopy: no signal in RG2 wild type, condensation and aggregation of RedXPRT due to DsRed tetramerization with subsequent XPRT lysosomal targeting in RG2/RedXPRT; less aggregation of RedXPRTΔ9c and Δ14c fusion protein is observed in the cell lines expressing C-terminal truncated forms. (B) FACS analysis of the corresponding samples in row A.
Statistics
The data obtained in the studies were analyzed using StatView software (SAS, Cary, NC). 95% Confidence intervals were identified for the numeric values and the standard deviations reported. Wilcoxon test was used for comparison of the paired values.
Results
In Vitro Expression of RedXPRT
Fluorescent microscopy. Revealed heterogenous levels of reporter gene expression (different intensity of fluorescence of individual cells) in a neomycin-selected population of RG2/RedXPRT (Figure 3). Several single-cell-derived clones of RG2/RedXPRT were obtained from the bulk culture, and these clones demonstrated a wide range of RedXPRT transgene expression levels. Clones with very high levels of RedXPRT transgene expression demonstrated significant intracellular condensation and compartmentalization of the fusion protein, resembling the pattern of lysosome distribution. These high-expressing clones exhibited slower growth rate as compared to those expressing intermediate or low levels of RedXPRT. Furthermore, the bulk-selected population of RG2/RedXPRT cells gradually became depleted of the cells expressing high levels of RedXPRT. These observations are consistent with previous reports of cellular toxicity associated with dsRed protein [40,41]. None of the clones expressing high levels of RedXPRT could be expanded to obtain the cell numbers sufficient for in vitro assessment of radiolabeled xanthine accumulation.
In vitro radiotracer accumulation studies. The rate constant for net accumulation (Ki, mL media/min/g cells) of [14C]-xanthine, 8-Br-[14C]-xanthine, and [3H]-hypoxanthine in wild-type RG2 and transduced RG2/RedXPRT cells are summarized in Table 1. A significantly higher rate of [14C]-xanthine accumulation was observed in the bulk-selected population of RG2/RedXPRT cells than that in wild-type RG2 cells (Figure 4A). Pretreatment of the cell cultures with allopurinol for 24 hr resulted in a doubling of [14C]-xanthine accumulation, Ki, in RG2/RedXPRT cells. In contrast, a twofold decrease in the rate of [14C]-xanthine accumulation was observed in allopurinol-treated wild-type RG2 cells (Table 1).
Unidirectional Accumulation Rates of Different Radiotracers in Wild-Type RG2 and RG2/RedXPRT Cells

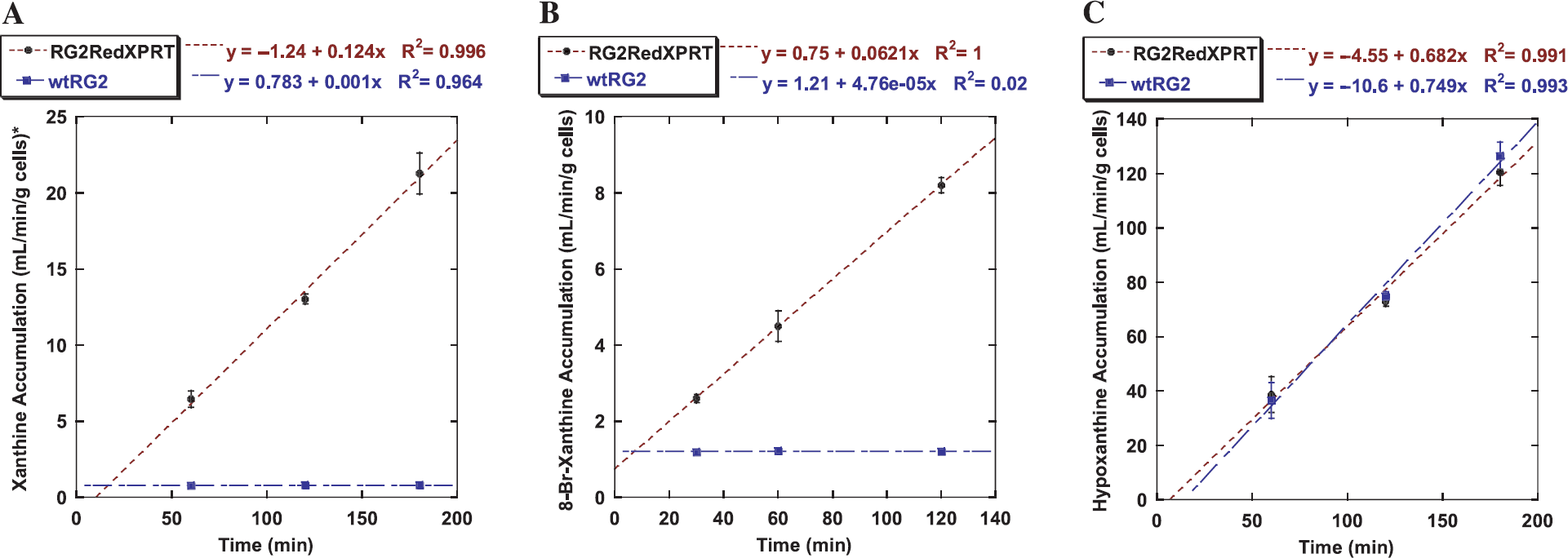
In vitro analysis of radiotracer accumulation in RG2/RedXPRT (brown dotted line) versus RG2 wild type (blue broken line). (A) [14C]-Xanthine accumulation. (B) 8-Br-[14C]-xanthine accumulation. (C) [3H]-Hypoxanthine accumulation. A significant accumulation of xanthine and 8-Br-xanthine is observed in RedXPRT-transduced cells, as compared to a mere equilibration of these tracers in the wild-type cells (A, B). In contrast, no difference in accumulation was detected for hypoxanthine (C).
8-Br-[14C]-xanthine accumulation in RG2/RedXPRT cells was similar than that of [14C]-xanthine, but it was sixfold lower than [14C]-xanthine in wild-type RG2 cells (Figure 4B), resulting in substantially higher specificity of 8-Br-[14C]-xanthine. In contrast, a very rapid accumulation of [3H]-hypoxanthine was observed in both wild-type RG2 and transduced RG2/RedXPRT cells (Figure 4C).
Impact of the C-Terminal Deletion in the XPRT Domain on the Subcellular Localization and Function of the RedXPRT Fusion Protein In Vitro
The RedXPRT fusion protein containing the wild-type XPRT sequence formed visible intracellular aggregates with a lysosomal pattern of subcellular distribution (Figure 3A). In contrast, both the short and longer truncations of the lysosomal-targeting motif (9 or 14 terminal amino acids, respectively) from the C-terminus of XPRT domain resulted in a more homogenous cytoplasmic distribution of the C-truncated RedXPRTA fusion protein (Figure 3A). The observed changes in subcellular distribution of RedXPRT did not affect the level of the transgene expression, as measured by FACS (Figure 3B). However, both the short and longer C-terminus truncations of the RedXPRTA fusion proteins resulted in a complete abolishment of their XPRT and hypoxanthine phosphoribosyltransferase activity (data not shown).
Assessment of Xanthine Lipohilicity
The octanol/water PC for [14C]-xanthine measured at pH 7.4 was 0.20 ± 0.001, equivalent to a logPoctanol of −0.69. This experimental value reasonably matched the values obtained from ChemDraw Ultra software, which were based on the molecular structure of xanthine: −0.27 ± 0.47 (by Crippen's fragmentation analysis) and −0.15 ± 0.49 (by Viswanadhan's fragmentation analysis). Estimates of the PS product of brain capillaries for xanthine based on the logPoctanol data, and molecular fragmentation estimates were 0.28, 0.75, and 0.99 mL/min/g brain, respectively, using the relationship described by Equation 1.
Xanthine Permeation of Brain Capillaries
The in vivo assessment of xanthine permeation of brain capillaries was performed using a double-tracer approach where [3H]-xanthine was compared to [14C]-iodoantipyrine in the same animals. An experimental paradigm to measure CBF in an anesthetized mouse was used [36]. The paired measurements of blood flow yielded values of 0.91 ± 0.17 (n = 6) and 1.04 ± 0.25 mL/min/g for [14C]-iodoantipyrine and [3H]-xanthine, respectively, in brain gray matter. Corresponding values in brain white matter were 0.62 ± 0.17 and 0.72 ± 0.24 mL/min/g, respectively.
In Vivo Expression of dsRed/XPRT
Subcutaneous tumors. Quantitative autoradiographic images (Figure 5C) of cryosections demonstrated highly selective accumulation of the [14C]-xanthine in the areas corresponding to the RedXPRT tumor tissue detected as basophilic on the H&E staining (Figure 5B). The maximum level of the radiotracer accumulation in the subcutaneous tumor was 3.2 ± 0.4% dose/g (the average was 2.4 ± 0.3% dose/g). The level of [14C]-xanthine accumulation in the control RG2/TKGFP subcutaneous tumors was low and similar to the muscle (0.78 ± 0.02% dose/g and 0.64 ± 0.05% dose/g, respectively). The high level of [14C]-xanthine accumulation spatially correlated with the fluorescence microscopy of adjacent frozen tissue sections. There was bright red fluorescence of the RG2/RedXPRT tumor without any significant 585 nm emission in the adjacent muscle tissue.
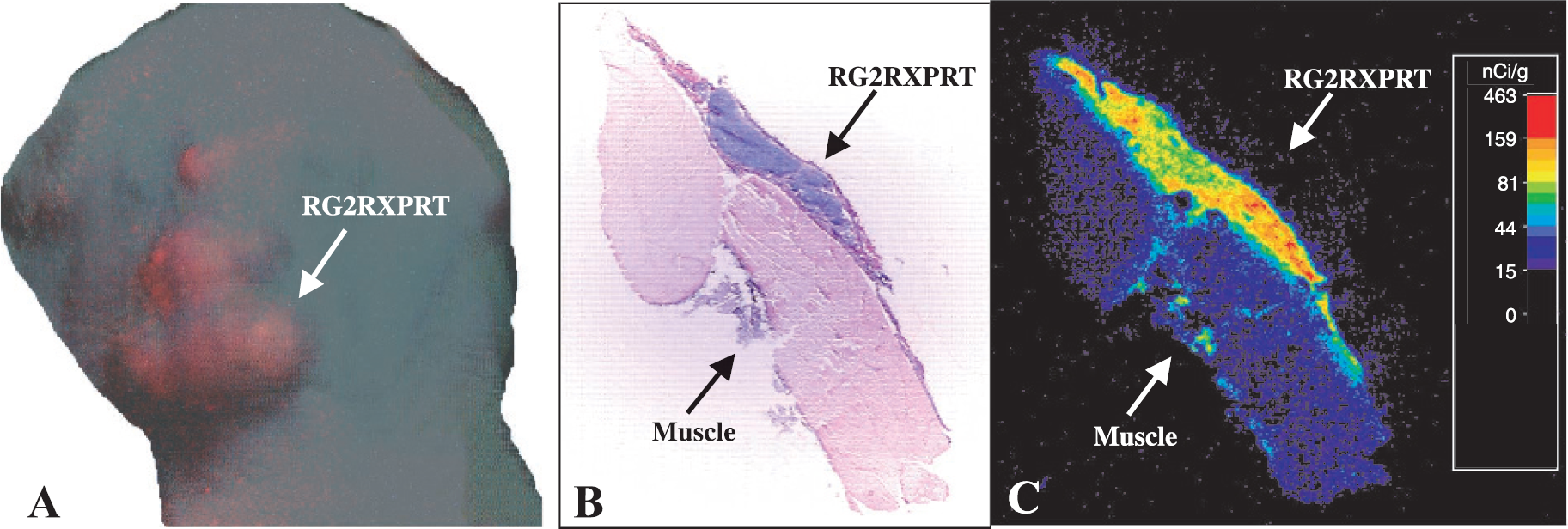
[14C]-Xanthine autoradiographic and fluorescent imaging of RG2dsRed/XPRT subcutaneous tumor (A) Whole-body transcutaneous in vivo fluorescent imaging of Red/XPRT reporter protein. (B) Hematoxylin/eosin stain of 20-μm cryosections. (C) Quantitative autoradigram.
The whole-body transcutaneous fluorescence imaging of the subcutaneous tumors allowed for registration of in vivo red fluorescence in the RG2/RedXPRT tumors (Figure 5A). The control RG2TK/GFP tumor was not producing any background fluorescence with 585 nm emission wave length.
Brain tumors. As described in the Materials and Methods, only those animals bearing intracerebral tumors that showed no signs of BBB disruption on Gd-DTPA-enhanced T1-weighted MRI were included into the current study (Figure 6C).
Quantitative autoradiographic studies demonstrated that [14C]-xanthine effectively crosses through the intact BBB and selectively accumulates to high levels (2.9 ± 0.3% dose/g) (Figure 6B), specifically in the areas infiltrated by RG2/RedXPRT cells (Figure 6A). In contrast, no specific accumulation of [14C]-xanthine was observed in the contralateral hemisphere, in the areas infiltrated by the RG2/TKGFP cells (Figure 6D). The level of [14C]-xanthine radioactivity in RG2/TKGFP intracerebral infiltrates was 0.64 ± 0.12% dose/g, which was only marginally higher than the surrounding brain tissue (0.36 ± 0.14% dose/g). Fluorescent in situ microscopy of the adjacent tissue sections (Figure 6E) demonstrated the localization of the RG2/RedXPRT tumor tissue in the area corresponding to radiotracer accumulation, which correlated with basophilic area of tumor tissue on the hematoxilin/eosin-stained slides (Figure 6A).

[14C]-Xanthine autoradiographic and fluorescent imaging of the brain tumors. Images obtained from 20-μm cryosections. RG2/TG—left hemisphere; RG2/RedXPRT—right hemisphere. Arrows point to the areas of tumor infiltration. (A) Hematoxylin/eosin-stained section of a rat brain. (B) Quantitative autoradigram. Squares delineate the areas of detail in the fluorescence microphotographs obtained from adjacent brain sections. (C) T1-weighted MR image of the animal head (axial projection). (D) eGFP fluorescent microscopy of RG2/TG tumor infiltrates of the brain. (E) dsRed fluorescent microscopy of RG2/RedXPRT area of infiltration in the contralateral hemisphere.
Unfortunately, deep localization, small size, and infiltrative pattern of growth of the brain RG2-dsRed/XPRT tumor made in vivo transcranial fluorescence imaging of intracerebral tumors impossible (data not shown).
Discussion
We have shown that the dual-modality reporter system, RedXPRT, can be used effectively for noninvasive imaging of reporter expression in the CNS with radiolabeled xanthine for nuclear imaging and for in situ fluorescence imaging. The use of multireporter gene technology for combined nuclear and optical imaging has several advantages. The coupling of a reporter gene for radionuclide imaging (e.g., HSV1-tk) with a reporter gene for optical imaging (e.g., eGFP) in fusion (TKGFP) [38] or in a single bicistronic transcription cassette [42] has previously been reported. More recently, a series of TKGFP mutants was developed with altered nuclear localization and better cellular enzymatic activity to optimize the sensitivity for imaging HSV1-tk/GFP reporter gene expression [43]. The RedXPRT fusion dual-reporter gene was constructed to provide an alternative dual-reporter system to TKGFP and to provide a better reporter gene for imaging within CNS structures. One segment of RedXPRT cDNA corresponds to the reporter gene for radiotracer imaging (KPRT), and the other segment corresponds to the reporter gene for optical imaging (dsRed). We have shown that the gene product, a fusion protein, retains the functional enzymatic (radiotracer imaging) and fluorescence (optical imaging) characteristics of XPRT and dsRed, respectively, although enzymatic activity is lost when the lysosomal-targeting motif is removed from the C-terminus of the XPRT domain.
E. coli XPRT was originally used as a selection gene to isolate transduced cells in vitro [44]. Also, it was demonstrated that E. coli XPRT could be used as a “suicide gene” in combination with the prodrug, 6-thioxanthine [45]. We selected E. coli XPRT as a suitable “reporter gene” because mammalian cells lack this enzyme and the XPRT enzyme is nontoxic to mammalian cells. Mammalian cells express hypoxanthine/guanine phosphoribosyltransferase (HGPRT), but xanthine is not a substrate for mammalian HGPRT. In contrast, radiolabeled xanthine and xanthine analogues are good substrates for bacterial XPRT (Figure 7). In the current work, we demonstrated that XPRT enhances the accumulation of radiolabeled xanthine and its analogue 8-Br-xanthine in transduced cells but not in wild-type mammalian cells. Using radiolabeled xanthine or its radiolabeled analogues, we expect to achieve: (1) high selective affinity to XPRT (as compared to endogenous mammalian HGPRT); (2) resistance to (or low) metabolic degradation, or the ability to block xanthine catabolism with allopurinol in order to (3) prevent the development of radiolabeled catabolites in nontransduced tissues; and (4) retention of the ribophosphorylated product in the tissue during the period of imaging.
The mechanism of metabolic entrapment of xanthine and its analogues by XPRT-transduced cells is somewhat different from the entrapment of FIAU in HSV1-tk-transduced cells. In HSV1-tk-transduced cells, FIAU crosses the cell membrane and is selectively phosphorylated by the HSV1 thymidine kinase. The reaction product, FIAU-monophosphate (FIAU-MP), is very polar, cannot cross the cell membrane, and is “metabolically entrapped.” In proliferating cancer cells, FIAU-MP can be phosphorylated by the cellular di- and triphosphate kinases to FIAU-TP, and effectively incorporated into the DNA (Figure 8A and B). However, cellular proliferation and the incorporation of FIAU into DNA is not the prerequisite for the successful entrapment of this radiotracer inside the HSV1-tk-transduced cells that generates the imaging signal.

Metabolic pathways of purine nucleotides. (A) Mammalian: hypoxanthine and guanine are the only possible substrate for mammalian purine nucleotide salvage pathway. (B) Bacterial, xanthine is utilized as well as hypoxanthine due to XPRT-mediated ribophosphorylation.
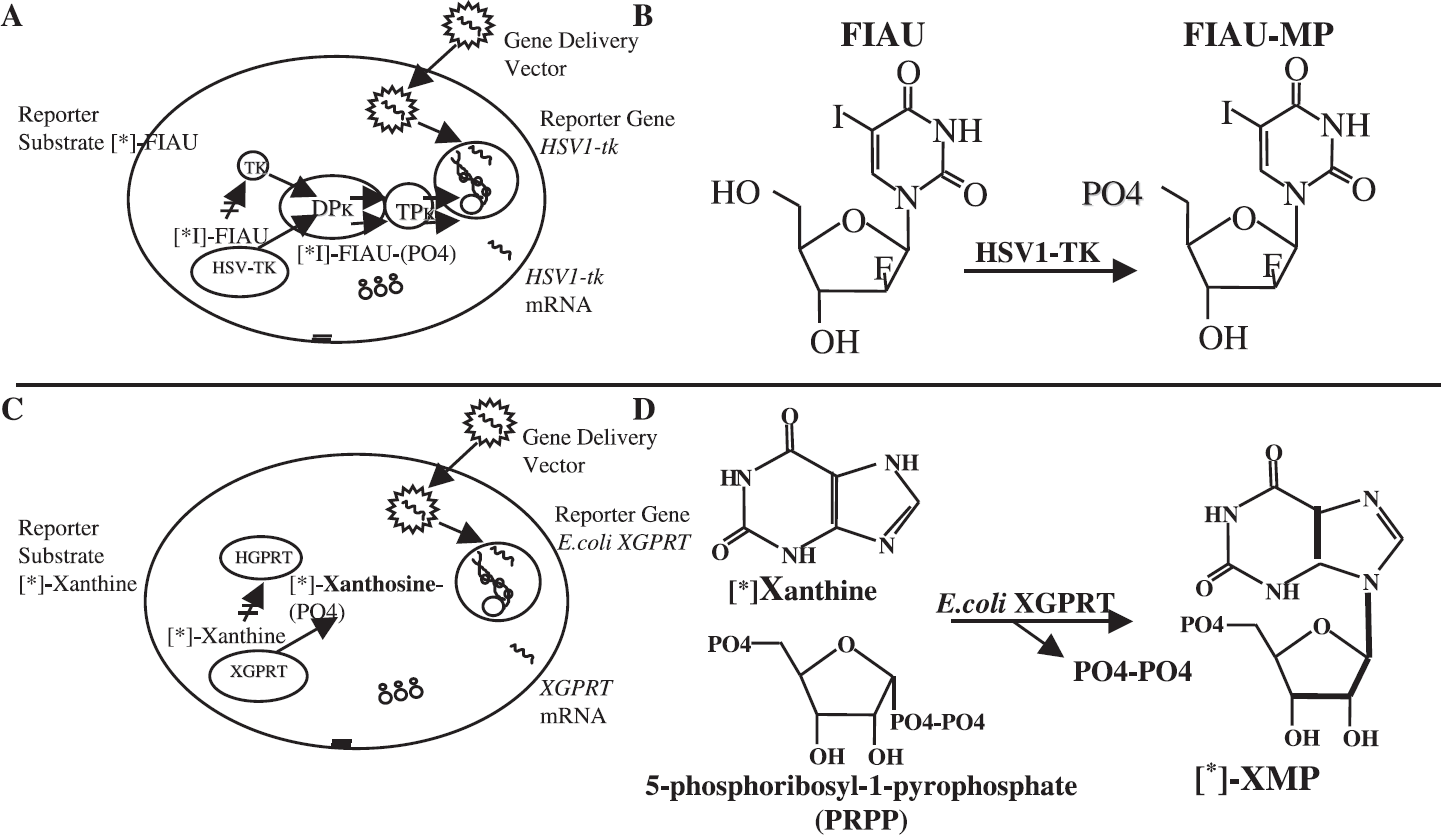
Differences in the mechanisms of the metabolic entrapment of FIAU by HSV1-tk and xanthine by E.coli XPRT. (A) HSV1-tk reporter gene paradigm. (B) Reaction of HSV1-tk-catalyzed [*I]-FIAU phosphorylation. (C) E. coli XPRT reporter gene paradigm. (D) Reaction of E. coli XPRT-catalyzed 8-[*]-xanthine ribophosphorylation.
In the case of E. coli XPRT, a somewhat different mechanism of substrate metabolic entrapment does not involve direct phosphorylation. In XPRT-transduced cells, xanthine (or an analogue) crosses the cell membrane and is phosphoribosylated by the XPRT. XPRT catalyzes the reaction between xanthine (or xanthine analogue) and 5-phosphoribosyl-1-pyrophosphate (PRPP) and forms the N-glucosidic bond of the reaction product, XMP. XMP cannot cross the cell membrane, and, thus, XMP is metabolically entrapped. The “second” (nonradiolabeled) substrate PRPP in the XPRT catalyzed reaction (Figure 8C and D) is ubiquitously present in the cell and is produced by PRPP synthase. Therefore, the XPRT-catalyzed reaction is not limited by PRPP availability (concentration), nor is it expected to be affected by the cell cycle or rate of cell proliferation [46,47]. Thus, the rate of radiolabeled xanthine (or xanthine analogue) ribophosphorylation and entrapment will depend on the level of XPRT expression in transduced cells.
Different xanthine analogues (e.g., 8-F-xanthine, 8-Br-xanthine) could be used to produce corresponding nucleotide monophosphates that are resistant to metabolic degradation by the mammalian 5′-nucleotidases and nucleoside phosphorylases [48]. The intracellular metabolic stability of the corresponding nucleotide monophosphates improves signal specificity and simplifies image interpretation due to the absence of radiolabeled metabolites. Xanthine also crosses the BBB at a sufficient rate such that it can be used to measure CBF, although the PS product estimated for rodent brain capillaries on the basis of xanthine lipophilicity was somewhat lower than the measured values for CBF. Nevertheless, kinetic modeling based on irreversible trapping of the XPRT reaction product, XMP, shows that the steady-state net accumulation rate, Ki, of the probe (e.g., xanthine) accurately reflects enzymatic activity (e.g., k3 for XPRT), provided k2 > > k3, where K1 and k2 are the influx and efflux constants of the probe, respectively, and Ki = (K1k3)/(k2 + k3) [49]. Under these conditions, probe delivery as well as vascular and cell membrane permeability is not limiting. Given a brain distribution volume (K1/k2) of about 1.0 mL/g for xanthine [50] and an influx constant of 0.5-1.0 mL/min/g (as reflected in the blood flow measurements), BBB equilibration (K1 and k2) will not significantly impact on the Ki measure for values of k3 that are 0.05 min−1 or less. Under these conditions, the measured Ki will be >90% of the Ki calculated using the same k3 and assuming infinite probe equilibration between blood and intracellular water (e.g., Ki = K1k3/k2).
Because xanthine readily penetrates the intact BBB, it is an ideal tracer for molecular imaging of the brain, including infiltrative brain tumor processes and micro-metastasis. To test the paradigm of effective molecular reporter gene imaging behind the intact BBB barrier, a biological model of transgene expression in the brain had to be selected. We used a rodent brain tumor model with known low BBB permeability RG2 [9,24,25], rather than a transgenic animal model with conditional expression of XPRT under a neuronal tissue specific promoter (i.e., GFAP). The RG2 cell line was transduced with a retroviral vector for a stable expression under a constitutive promoter, RG2/RedXPRT, and was used for the in vitro and in vivo studies reported here. Tumors were studied when they were small and infiltrative, prior to BBB disruption, and this was confirmed by contrast MRI performed just before the radiotracer studies.
The development and radiolabeling of xanthine analogs deserves consideration, particularly in light of the positive in vitro results that were obtained with 8-bromo-xanthine. Cyclic bromo-AMP is resistant to degradation, as Br stabilizes the C-N bond [51], and 8-chloro-xanthine and other halogen derivatives of the nucleotide are potent inhibitors of XO [51]. Unfortunately, placement of a fluorine instead of a bromine in the 8-position is likely to produce an unstable molecule, in contrast to what was previously demonstrated with ganciclovir [16]. Labeling of xanthine with 11C-methyl in the 8-position is not feasible, because it will eliminate the affinity to XPRT (see Appendix).
It is noteworthy that our mutagenesis studies aimed to minimize the lysosomal targeting of XPRT and improve its cytoplasmic distribution in fusion with dsRed protein yielded a complete abolishment of metabolic activity in C-terminus-truncated XPRT mutants. This phenomenon could be explained in part by the propensity of the 12 amino acid residues at the C-terminus of the E. coli XPRT subunit to adopt a random coil conformation (Figure 9A) that resembles an arm extending away from the subunit (a feature that is not observed in other PRTase structures). The “arm” extends across the tetrameric E. coli XPRT complex to interact with Ser-A26 of a crystallographically related dimeric subunit. The interaction between Arg-C153 and Ser-A26 could be important in context of substrate binding and/or catalysis and could also explain the requirement of E. coli XPRT tetramer formation for catalytic activity [89].
The best-developed reporter system for nuclear-based imaging, involving the herpes virus type one thymidine kinase (HSV1-tk) and radiolabeled nucleotides [1], does not provide a sufficient solution for reporter imaging in the CNS with an intact BBB. Although HSV1-tk gene-based reporter systems have used different radiolabeled nucleosides as reporter probes, including FIAU [1,38]–[52,43], FGCV [54], FPCV [54], FHPG [53]–[56], and FHBG [5,53,56], these nucleosides and nucleoside analogues do not effectively cross the intact BBB [57]–[59]. However, [131I]- and [124I]-IUdR have been shown to accumulate in the brain tumor tissue that does not contrast-enhance on Tl-weighted MRI after Gd-DTPA administration [60,61], and [*]-FIAU does accumulate in HSV1-tk-expressing tumors with a compromised BBB. This has been demonstrated in both an animal model [1] and in patients [62]. Nevertheless, the level of FIAU accumulation in HSV1-tk-expressing brain tumors could be influenced by heterogeneity in local microvascular permeability. The application of the HSV1-tk-based reporter imaging has limited applicability to studies in CNS; namely, for pathological processes associated with an impaired BBB.
The hD2R [63,64] and the human somatostatin receptor subtype-2 (hSSTR2) [65,66] genes have been suggested as potential reporter genes for human studies. Both human genes have limited expression in the body; hD2R expression is limited to the striatalnigral system of the brain and high hSSTR2 expression is largely limited to carcinoid tumors. This approach is a very clever strategy because there are established complimentary radiolabeled probes for each of these reporter genes; 3-(2′-[18F]fluoroethyl)spiperone (FESP) for hD2R imaging [67], and [111In] DTPA-octreotide (a complimentary radiolabeled somatostatin analogue) for hSSTR2 imaging [68]. Furthermore, both probes are approved for human administration. Both of these reporter systems have distinct benefits with respect to initiating molecular/reporter imaging in human subjects. However, receptor expression on the external surface of cells or on specific intracellular organelles is a complex process. Receptor expression involves intracellular trafficking and cell membrane incorporation that is likely to be altered under different conditions and different disease states. Furthermore, cell surface receptor expression may account for only a small part of total receptor protein produced by transcriptional activation of the reporter gene. It remains to be shown whether imaging receptor-based reporter systems (e.g., the hD2R and hSSTR2 reporter gene systems) will provide a consistent and reliable measure of reporter gene expression under variable stress or altered conditions.

dsRed/XPRT stereochemical reconstruction. (A) Dimeric structure of E. coli XPRT. The XPRT crystals grown in the absence of substrate or product were used to determine the structure of XPRT at a resolution of 1.8 A by multiple isomorphous replacement [91]. Superimposition of the human HPRT/GMP complex (dark shading) with the structure of E.coli XPRT (light shading) in the base-specifying region of human HPRT.
The hD2R-[18F]-FESP system [16,17] is attractive because [18F]-FESP does penetrate the BBB and is selectively accumulated in M)27?-expressing cells. This is a well-established system that has been used to study D2 receptors is patients. However, D2 receptors are highly expressed on dopaminergic neurons in the caudate nucleus, and this could significantly impact on the specificity of reporter imaging in the brain with [18F]-FESP. The data regarding permeability of the BBB by different somatostatin analogues are rather controversial. The original studies on permeability of octreotide peptides through the BBB demonstrated that these peptides have very limited ability to cross the intact BBB [69]. It was demonstrated that BBB permeability of radiolabeled octreotide analogues does not differ from that of the DTPA [70], and the results of other studies suggested a paracellular route of octreotide transport and a minimal contribution of carrier-mediated transport mechanisms [71]. More recently, active transcapillary efflux of octreotide in the brain was demonstrated through involvement of p-glycoprotein and Mrp2 [72]. These receptor-based reporter gene approaches have significant limitations for imaging in the CNS due to highly specific accumulation of reporter probes in certain structures of the brain that do not express the reporter transgene, or due to their poor BBB permeability.
Multimodality reporter gene imaging combining nuclear and optical options was first developed in our laboratory to provide expanded imaging and analytic capability [38]. The fluorescence component facilitates and lowers the cost of the development and validation of new reporter constructs (Table 2). This is accomplished by FACS analysis and by in vitro florescence assays. Low-cost in vivo fluorescence imaging can be performed repetitively on the same animal, prior to performing more expensive gamma camera or PET imaging. In addition, the in vivo fluorescence images of reporter gene expression can be directly compared to the corresponding PET images. Quantitative autoradiographic images can also be directly compared to corresponding in situ fluorescence and histological images from the same or adjacent tissue sections. In vivo imaging of optical reporter genes can provide real-time data that indicate the spatial distribution of tumor cells at multiple time points during the course of disease [5,6,73]–[82].
Optical reporter gene imaging provides qualitative (or semiquantitative) information with relatively short image acquisition times. This facilitates rapid assessment for potential interventions or for obtaining a more expensive and quantitative nuclear scan [75,81,82]. Because groups of animals can be followed over time, far fewer animal subjects are required to obtain statistically meaningful results. The stress on the animals in these studies is dramatically reduced because whole-body imaging replaces death as an endpoint and the models can be evaluated in minimal disease states where the tumor burden is small. Rapid whole-body imaging and the use of repeated measurements on groups of animals accelerates the study, reduces the variablility of interanimal comparisons and improves the power of the statistical analysis. Coupling of fluorescent and radionuclide imaging provides improved spatial and quantitative assessments of reporter gene expression at the microscopic (fluorescent microscopy and autoradiography) as well as macroscopic (PET, gamma camera, transcutaneous fluorescence) level. Thus, dual-modality (nuclear and fluorescence) imaging is a very useful combination, and this has been confirmed in several recent studies [2,3,5]. It allows for a “seamless” transition from the in vitro—to in vivo—to in situ imaging technologies.
Multimodality Reporter Genes

The dual-reporter system containing dsRed and XPRT genes in fusion (RedXPRT) provides other benefits, in addition to being a better reporter for imaging in the CNS. It provides a second dual-reporter system that can be combined in novel ways with our established TKeGFP dual reporter [2,3,8]. One application is the combination of inducible and constitutive reporter systems in a single construct. This combination has already been tested in preliminary studies [83,84] and demonstrates the feasibility and utility of this construct. The constitutive component (e.g., CMV-driven RedXPRT) has been termed the “beacon element” and can be coupled with an inducible component (e.g., HIF-1α-driven TKeGFP) that has been termed the “sensor element.” This double reporter construct (containing two dual-reporter systems) is particularly useful for identifying transduced cells (constitutive-beacon reporter) and to assess whether these transduced cells are being activated (inducible-sensor reporter). This is particularly useful and necessary when in vivo transfections/transductions of reporter genes are performed in native tissue or in transgenic animals. In such cases, it is necessary to normalize the readout of the inducible-sensor reporter by the readout of the constitutive-beacon reporter. This accounts for the efficiency of transfection/transduction and for the number of transduced cells in each voxel element of the image. Such double reporter systems (with combined inducible and constitutive reporter readouts) will broaden the spectrum of molecular gene imaging studies and will provide the opportunity to study biological processes noninvasively in both animals and human subjects.
Conclusions
The newly developed dsRed/XPRT reporter gene-[*]xanthine reporter probe system provides a second nuclear and optical imaging technique for combined PET/QAR and fluorescent imaging applications. This system with [11C]-xanthine could be used for rapid repetitive PET imaging of different molecular-genetic processes. The dsRed/XPRT-xanthine approach is better suited for reporter imaging in the CNS in comparison to other currently available reporter systems, particularly if the BBB is intact. Xanthine readily crosses the BBB and probe delivery and brain-blood equilibration does not appear to be limiting. The RedXPRT dual reporter can also be combined with the TKeGFP dual reporter to obtain both an inducible and constitutive double reporter in the same vector construct. XPRT also has the potential for becoming a novel reporter gene that could be used in translation-patient studies in the future.
Appendix. Structure-Specificity Relationships of Other Potential Probes for Imaging XPRT Expression
The mammalian HGPRT (EC 2.4.2.8) catalyzes ribophosphorylation of guanine to produce guanosine monophosphate. Mammalian HGPRT also catalyzes ribophosphorylation of hypoxanthine to produce inosine monophosphate (IMP), which is converted into XMP by the IMP-dehydrogenase, and which, in turn, is converted into guanosine monophosphate (GMP). Mammalian cells do not utilize xanthine to produce XMP for synthesis of GMP because xanthine is not a substrate for mammalian HGPRT [85]–[87]. In contrast, E. coli XPRT (EC 2.4.2.22) catalyzes the reaction with xanthine where the reaction product, XMP, is utilized for GMP synthesis [44].
Structure-Activity Relationships
XPRT from E. coli is a tetrameric enzyme having 152 residues per subunit. XPRT catalyzes the transfer of the phosphoribosyl group from 5-phospho-alpha-
Base Specificity
E. coli XPRT catalyzes the transfer of a phosphoribosyl group to 6-oxopurine bases, discriminating against 6-aminopurine bases, such as adenine. The kinetic constants for E. coli XPRT are summarized in Table 3-Comparison of kcat/Km values for the 6-oxopurine bases revealed that E. coli XPRT is able to utilize bases in the following order: guanine > xanthine ⋙ hypoxanthine. In contrast, the HPRTs from Tritrichomonas foetus and Toxoplasma gondii have high specificity for hypoxanthine and guanine but lower specificity for xanthine, while human HPRT has specificity for hypoxanthine and guanine only.
Kinetic Constants for E. coli XPRT
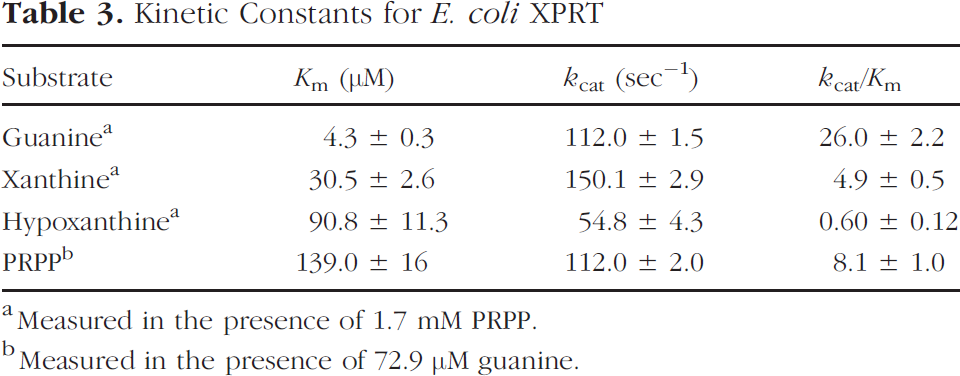
Measured in the presence of 1.7 mM PRPP.
Measured in the presence of 72.9 μ guanine.
The specificity for 6-oxopurines in the three HPRTs appears to be defined by three different interactions: (1) an interaction between a positively charged protein side chain (Lys-134 in T. foetus HPRT, Lys-178 in T. gondii HGXPRT, and Lys-165 in human HPRT) with the 6-oxo group on the purine base (thereby selecting against the 6-aminopurines); (2) a stacking interaction between an aromatic side chain (Phe-162 in T. foetus HPRT, Trp-199 in T. gondii HGXPRT, and Phe-186 in human HPRT) and the purine base; and (3) an interaction that defines the enzyme specificity for guanine, hypoxanthine, or xanthine (these bases have respectively an amino, a hydrogen, or a carbonyl oxygen atom at the 2-position of the 6-oxopurine base system). In the structure of the human HPRT/GMP complex, this third specificity interaction appears to be defined by the main chain carbonyl groups of Val-187 and Asp-193, which interact favorably with the 2-amino group of the guanine base to promote its interaction with the enzyme (Figure 9B). It was demonstrated that the same main chain oxygens would also allow space for the binding of hypoxanthine but would not favor the binding of the 2-oxo group of xanthine [88]. In contrast, in T. foetus HPRT, the side chain OH group of Tyr-156 interacts with the 2-amino group of guanine, and because this group can act both as a hydrogen bond donor and acceptor, this residue could form a hydrogen bond to either the guanine amino group or the xanthine carbonyl oxygen [89]. In T. gondii HGXPRT, the mechanism for base discrimination in not yet clear, but it appears that the backbone conformation differs sufficiently from that of human HPRT to allow xanthine to bind the enzyme [90].
Comparison of human HPRT/GMP complex with that of the E. coli XPRT allows the identification of residues that could define the binding specificity of E. coli XPRT for 6-oxopurines (Figure 9B). It was suggested that the positively charged residue in E. coli XPRT that selects for 6-oxopurines (and against 6-aminopurines) is probably Lys-115, which occupies a similar position to Lys-165 of human HPRT. The residue in E. coli XPRT equivalent to the aromatic residue that interacts with purine base in human HPRT (Phe-186) and T. gondii HGXPRT (Trp-199) was suggested to be Trp-134, which can stabilize the purine ring system [91].
Also, it was suggested that Gln-137 could be the residue in E. coli XPRT, which discriminates between three 6-oxopurine bases (Figure 9B). Thus, binding of guanine could be stabilized by E. coli XPRT through interaction of the 2-amino group of the substrate with Gln-137 Oɛ2. Similarly, binding of xanthine could be stabilized through interaction of its 2-oxo group with Gln-137 Nɛ2. The lower affinity of E. coli XPRT to hypoxanthine, which has no exocyclic group at the 2-position, might be explained by the loss of interaction with Gln-137 compared with other 6-oxopurines. Therefore, it was suggested that the specificity of E. coli XPRT for guanine and xanthine does not appear to be due to the main chain oxygen interactions seen in some of the HPRTs, but rather may be uniquely determined through interaction with a Gln-137 residue [91].
Additional studies [92] have demonstrated that the purine base specificity of E. coli XPRT appears to be due to water-mediated interactions between the 2-exocyclic groups of guanine or xanthine and side chains of Glu 136 and Asp 140, as well as the main-chain oxygen atom of Ile135. Asp92, together with Lys115, could help stabilize the N7-protonated tautomer of the incoming base and could act as a general base to remove the proton from N7 when the nucleotide product is formed. The 2.6 A resolution structure of E. coli XPRT complexed with product GMP is similar to the substrate-bound complexes. However, the ribose ring of GMP is rotated by approximately 24° compared with the equivalent ring in cPRib-PP. This rotation results in the loss of all interactions between the ribosyl group and the enzyme in the product complex [92].
Several xanthine analogues (e.g., 8-F-xanthine, 8-Br-xanthine) have the potential to produce corresponding nucleotide monophosphates resistant to catabolism by the mammalian 5′-nucleotidases and nucleoside phosphorylases [48], which contributes to signal specificity and facilitates image interpretation reducing the background of radiolabeled metabolites. Unfortunately, to date, there are no reports on structure-activity relationships for the binding of ligands to E. coli XPRT. However, as we have described, there is a significant structural similarity between the E. coli XPRT and T. gondii HGXPRT (Figure 10). Therefore, the data obtained by Naguib et al. [93] on structure-activity relationships for the binding ligands to T. gondii HGXPRT may be applicable and transferable to some extent to E. coli XPRT.
A pyrrole-nitrogen (NH) appears to be strongly preferred or required for binding, as the presence of an exocyclic methyl group at the N1 position decreases binding of guanine and xanthine 15- and 230-fold, respectively. Exocyclic substitutions on the 2-position of purine generally increase binding (oxo is preferred over amino and thio substitutions) to the enzyme (Glu136, Asp140, and Ile135 of the E. coli XPRT binding pocket). Exocyclic substitutions at N3 position decrease or even abolish binding. The loss or decrease in binding appears to be a direct result of steric hinderance (e.g., xanthine-N3-oxide, 3-methylxanthine). It was demonstrated that an unsaturated pyrrole-type nitrogen (NH) or a pyridine-type nitrogen (N-) at the 3-position is required for binding.
In the C6 position, the oxo group is preferred over a thio substitution. A 2,6-dithio substitution results in a further decrease of binding. Halogeno substitution in the C6 position by CI (6-cloropurine) results in a weaker binding as compared to a 6-thio substitution; a 6-iodo substitution (6-iodopurine) completely inhibited binding to the enzyme. Although the chloro group is more electronegative than the iodo group, the iodo (2.2 Å) is larger than the chloro (1.8 Å) group. It appears, that the active site of the enzyme, which accomodates C6 substitutions (Lys 115 and Lys 178 of the E. coli and T. gondii enzymes, respectively), is more sensitive to size than the electronic characteristics of the substituent (i.e., electron-withdrawing or electron-donating). This is further confirmed by the decrease in binding of 6-methoxypurine, 6-methylaminopurine, 6-benzylaminopurine, and 6-methylpurine. Substitutions of hydrogen in the N7 position by an exocyclic methine group decreases binding by more than 10-fold, whereas 7-methyl substitution decreases binding more than 100-fold, suggesting that only a hydrogen atom or a nitrogen with an unshared pair of electrons can reside at the 7-position.
A methine group (C-) at the 8-position is required for binding to the enzyme. Replacement of the 8-position methine of hypoxanthine, guanine, or xanthine with a nitrogen, to yield 8-azahypoxanthine, 8-asaguanine, and 8-azaxanthine, respectively, significantly reduces (>400-fold) or abolishes binding to the enzyme. The N8 nitrogen of 8-azapurines exists predominantly as “pyridine-type” nitrogen (N-) that has an unshared pair of electrons that are not involved in the aromaticity of the heterocycle. Therefore, poor binding of the 8-azapurines may be due to the electronic influence of the 8-position nitrogen on hydrogen bonding at N7 or possibly charge repulsion within the catalytic site of the enzyme (presumably Lys 115 and Asp-92 of the E. coli XPRT; see Figure 4). Substitutions in C8 position of hypoxanthine with an oxo (8-oxohypoxanthine) or thio group (8-thiohypoxanthine) significantly reduced binding. Thus, the size of the substituents at the C8 position definitely affects the affinity of the analogue to the enzyme.
9-Position endocyclic substitutions of N9 with a methine group does not affect binding. However, any exocyclic substitutions in the 9-position larger than a hydrogen (e.g., 9-methylguanine) would preclude binding the enzyme due to overlap with the PRPP binding site.
There are no data on the effects of 8-halogeno substitutions (F, Cl, Br, I) of on binding of xanthine, hypoxanthine, or guanine to the E. coli or T. gondii HGXPRT enzymes, except for 8-bromoguanine and T. gondii XGPRT. It was demonstrated that 8-bromo substitution at C8 of guanine (8-bromoguanine) decreased binding to the T. gondii HGXPRT [93]. Our data comparing xanthine and 8-bromoxanthine confirm this structure-activity observation (see Figure 4). Based on a comparison of the 8-aza, 8-oxo, 8-thio, and 8-bromo substitutions, the 8-fluoro substitution is expected to have low steric influence on the purine base conformation (specifically in Nl). While retaining the small size of the 8-position group (similar to that of the hydrogen 1.2 Å), the 8-fluoro substitution (fluorine 1.35 Å) is expected to increase electronegativity of the 8-position, and result in an increased binding to the enzyme (E. coli XPRT). Similar observations were by Barrio et al. [94] who reported a sixfold increase in 8-fluoroacycloguanine binding to herpes viral thymidine kinase over guanine, and 10% increase in 8-fluoroadenine binding to adenosine deaminase over adenosine [95].

Alignment of the amino acid sequences of E. coli XPRT and T. gondii HPRT. Top line: E. coli XPRT; lower line: T. gondii HPRT. Highly conservative regions forming the allosteric center are outlined in bold and color.
In general, the choice of marker substrates should be based on a combination of several factors that are important for in vivo imaging. These factors include: (1) resistance to metabolic degradation by the host enzymes; (2) rapid passage through the capillary wall and cell membranes (either by diffusion or by facilitated transport); (3) the half-time elimination kinetics from blood that would be compatible with the kinetics of substrate accumulation in transduced tissues (longer half-time elimination kinetics from blood is required to facilitate slow accumulation of substrate in transduced tissues, and vise versa); (4) the rate of substrate accumulation in transduced tissue that produces levels of accumulated radiolabeled product that are adequate for noninvasive imaging; (5) dosimetry; and (6) complicity of synthesis and/or radiolabeling. Sometimes, a compromise for the affinity of a marker substrate to a marker enzyme is must be made to accommodate other factors (listed above) that impact on successful implementation of imaging paradigm in clinical settings. Therefore, the choice of marker substrates for imaging E. coli XPRT expression cannot be solely based on structure-activity relationships observed (or predicted) for the purified enzyme (with an optimized reaction buffer) in a cell-free system. Such choice should be based on the results of studies on substrate accumulation in cell cultures and pharmacokinetic, pharmacodynamic, and imaging studies in animals.
In the XPRT-xanthine analogue reporter gene-reporter probe approach, the 8-, 6-, and 2-position-modified radiolabeled analogues of xanthine could potentially be used for imaging XPRT expression, based on previously reported data on their affinity to XPRT. These compounds inhibit several enzymes involved in metabolic pathways of DNA and RNA synthesis and are only minimally (if not at all) ribophosphorylated by mammalian HGPRT. These xanthine analogues can be radiolabeled in 8-position with several different radionuclides, including [18F] and [76Br], appropriate for clinical imaging with PET. The radiolabeling is relatively easy and could be performed in most nuclear medicine PET facilities. The longer half-life of the [Br] provides an additional advantage with respect to production and distribution of radiotracers from a central source.
An important characteristic of the 8-halogeno substitutions of xanthine is that it provides a protective group against metabolic degradation in blood and tissues. The first step in the metabolic degradation of xanthine is oxidation by XO, which results in production of uric acid [96]. This oxidation reaction is essentially abolished by the 8-halogeno substitutions of xanthine. For example, the 8-Br-xanthine is not oxidized, but it is an inhibitor of XO with a Ki of approximately 400 μM. Inhibition is noncompetitive with respect of xanthine and noncompetitive with respect to molecular oxygen (binding at the active site of the enzyme that contains the molybdenum center) [97]. Also, it is important to note that XO could be effectively inhibited by allopurinol, a drug commonly used to treat gout since the 1960s [98].
The 8-halogeno-substituted analogues of xanthine are expected to be excreted largely unchanged in the urine. As a result, a greater amount of nondegraded radiolabeled drug may be delivered to the target tissues and confounding problems associated with imaging radiolabeled metabolites in both target and surrounding tissue should avoided. The 8-halogeno substitutions on xanthine moiety should further decrease the affinity to mammalian HGPRT while minimally affecting its affinity to E. coli XGPRT, and thus would increase “selectivity” towards XGPRT. In addition, the 8-halogeno substitutions on xanthine moiety protects from dephosphorylation and enzymatic cleavage of the N-glucosidic bond of the resulting reaction product, 8-halogeno-xanthosine, by nucleoside phosphorylases. The increased stability of the 8-halogeno-XMP results in a significant prolongation of intracellular half-life of the accumulated tracer (more stable intracellular retention of accumulated radioactivity) and simplifies interpretation of images. Due to their metabolic stability, the 8-halogeno-substituted purine nucleotides (e.g., 8-Br-cAMP) are routinely used in signal transduction research [48,99]–[101]. Recently, Barrio et al. demonstrated that the 8-fluoro substitution in 8-fluoroguanine and 8-fluoroacycloguanine results in an increased metabolic stability of these nucleosides to metabolic degradation [94].
Thus, 8-halogeno analogues of xanthine should (may) have specific advantages over xanthine as “marker substrates” for imaging XGPRT expression. This is primarily due to: (1) higher selective affinity to XGPRT (as compared to endogenous mammalian HGPRT); (2) improved resistance to metabolic degradation by nucleoside phosphorylases; (3) very low (or absence) of radiolabeled metabolites in non transduced tissue; and (4) metabolic entrapment and retention of the reaction product in transduced tissue during the period of imaging. These four advantages eliminate (or markedly reduce) the problems associated with imaging radiolabeled metabolites in both target and surrounding tissue, and they facilitate quantitative measurements and interpretation of the resultant images.
Comparison of Structure-Specificity Relationship of Different Purine Analogues with the Best Apparent Ki values for Inhibition of T. gondii XGHPRT (Selected Data from Naguib et al. [93])

aSources indicated: Sigma, St. Louis, MO (SIG), Nutritional Biochemicals, Cleveland, OH (NBC), Chemical Dynamics, South Plainfield, NJ (CDC).
Measured with 10 μM of xanthine.
Measured with 4 μM of guanine.
The reason for pursuing studies with 18F- and 76Br-labeled analogues is the wider availability and substantially lower cost of labeling with 18F for PET imaging, and the ability to perform repeat imaging studies of gene expression over time. Labeling with 76Br (t1/2 = 16.3 h) is also feasible for PET imaging; it would allow for central production facilities and longer washout times to allow for tracer accumulation in transduced tissues to the levels that are adequate for imaging, and it would permit repeat imaging every 2–3 days.
Other xanthine analogues (2- and 6-position-substituted 8-halogenated analogues) may be even more selectively ribophosphorylated by the E. coli enzyme XGPRT and be more resistant to metabolic degradation than xanthine. Only limited data exist in the literature regarding structure-specificity relationships for XGPRT from E. coli. The most complete study on structure-specificity relationships for binding of various 2- and 6-position-substituted purine analogues is reported for XPRT of T. gondii [93]. These analogues were assessed as potential therapeutic agents for toxoplasmosis because they have little affinity to the mammalian HGPRT (Table 4).
Among these analogues (Table 4), we initiated studies with three analogues (in addition to xanthine) for 8-fluorination and 8-bromination because they are the most likely successful marker substrates of E. coli XPRT. These analogues include: 6-thiopurine (
Footnotes
Acknowledgments
This work was supported by NIH grants CA57599, CA76117, and CA83084, Department of Energy grant DE-FG02-02ER63481, and the DANA Foundation Award for Clinical Concepts in Neuro-Immuno Imaging.