
Abstract
The parasympathetic nervous system is likely to be involved in migraine pathogenesis. We hypothesized that the cholinomimetic agonist carbachol would induce headache and vasodilation of cephalic and radial arteries. Carbachol (3 μg/kg) or placebo was randomly infused into 12 healthy subjects in a double-blind crossover study. Headache was scored on a verbal rating scale from 0–10. Velocity in the middle cerebral artery (Vmca) and diameter of the superficial temporal artery (STA) and radial artery (RA) were recorded. Nine participants developed headache after carbachol compared with three after placebo. The area under the curve for headache was increased after carbachol compared with placebo both during infusion (0–30 min) (P = 0.042) and in the postinfusion period (30–90 min) (P = 0.027). Carbachol infusion caused a drop in Vmca (P = 0.003) and an increase in STA diameter (P = 0.006), but no increase in the RA diameter (P = 0.200). In conclusion, the study demonstrated that carbachol caused headache and dilation of cephalic arteries in healthy subjects.
Keywords
Introduction
The parasympathetic nervous system, which for a long time has been implicated in headache pathogenesis (1), is believed to contribute to the activation and sensitization of perivascular sensory nerve fibres, leading to migraine pain (2–4). Parasympathetic fibres innervate pain-sensitive intra- and extracranial arteries and contain various transmitters such as vasoactive intestinal polypeptide (VIP) (5–8), pituitary adenylate cyclase activating peptide (PACAP) (8) and acetylcholine (5–7). Human studies have shown that both PACAP38 and VIP are vasoactive, but with different headache-eliciting effects (9, 10). Fibres containing acetylcholine-producing enzymes are present in cerebral (6, 11), dural (5) and temporal (12) arteries in close relation to the advential-medial borders of the vessel wall. Acetylcholine dilates these arteries through muscarinic receptors located on the endothelium (5, 11, 12). Activation of these receptors leads to activation of nitric oxide synthase (NOS) (13) and thereby production of nitric oxide (NO), which is a well-known trigger of neurovascular headache (14). Furthermore, acetylcholine may directly excite sensory afferent fibres (15–17), and inhalation of the cholinergic agonist methacholine has previously been reported to induce headache in a case observation of one subject (18). Acetylcholine-induced headache, and its effect on brain haemodynamics in humans in vivo have not been systematically studied. Acetylcholine is quickly degraded by cholinesterases and therefore difficult to study in man. Carbachol is a cholinomimetic agent that is not metabolized by cholinesterases and therefore suitable for exogenous administration.
In this study we hypothesized that infusion of carbachol would induce headache in healthy subjects, and that this headache would be associated with dilation of cephalic and radial arteries. To test this hypothesis we performed a double-blind, placebo-controlled, crossover study exploring the headache and haemodynamic effect of carbachol.
Design and methods
Carbachol experimental model of headache
Pilot experiment
To determine the optimal dose of carbachol that decreased blood flow velocity of the middle cerebral artery (VMCA) and induced headache, we conducted an open-label pilot study. Four healthy subjects were included (one female and three male). The subjects received carbachol in stepwise increased doses of 2, 3 and 4 μg/kg. Each dose was given over 25 min. There was a wash-out period between each infusion lasting ≥ 60 min. One male subject was excluded after feeling dizzy during and after 2 μg/kg carbachol infusion. The subject was observed for 2 h and discharged without any complaints. The pilot study showed that a dosage of 3 μg/kg produced a mild headache and a decreased VMCA. At a dosage at 4 μg/kg the subjects experienced intolerable cholinergic side-effects, in particular a very intense and painful urge to void.
Main experiment
We recruited 12 healthy subjects (7 female and 5 male), mean age 23 years (range 20–29 years). Exclusion criteria were: a history of migraine or any other type of headache (except episodic tension-type headache < 5 days/month); any daily medication apart from oral contraceptives; serious somatic (including any history or sign of asthma) or psychiatric disease. The study was approved by the Ethics Committee of the County of Copenhagen (KA20060085) and the Danish Medicines Agency (EudraCT-nr: 200600246219). The study was monitored by Copenhagen University Hospital GCP-unit and registered at Clinicaltrials.gov (NCT00357864). All subjects gave informed consent to participate in the study, which was undertaken in accordance with the Helsinki Declaration of 1964, as revised in Edinburgh in 2000.
Experimental design
In a double-blind, placebo-controlled, crossover design, the subjects were randomly allocated to receive 3 μg/kg carbachol or placebo (isotonic saline) over 25 min on 2 days separated by a least 1 week. Before the experiment each subject underwent a general physical examination and measurement of serum thyroid-stimulating hormone, since carbachol can theoretically induce atrial fibrillation in hyperthyroid subjects.
All subjects reported to the laboratory 08.30 h headache free. The intake of coffee, tea, cocoa or other methylxanthine-containing foods or beverages was not allowed for the last 8 h before the start of the study. All procedures were performed in a quiet room at a temperature of 25 oC. Subjects were placed in the supine position and a venous catheter was inserted into the right antecubital vein for drug infusion. The subject then rested for at least 30 min before baseline measurements of blood pressure (BP), heart rate (HR) and ECG were performed, and the infusion started using a time- and volume-controlled infusion pump. Headache intensity, VMCA, superficial temporal artery (STA) and radial artery (RA) diameter, end-tidal partial pressure of CO2 (PetCO2), adverse events and vital signs were recorded at T –10, T 0 and then every 10 min until 90 min after the beginning of infusion. Cerebral blood flow (CBF) measurements by single-photon emission computed tomography (SPECT) were performed with six randomly allocated subjects. CBF was measured at T 0, T 20 and T 60. The subjects were discharged from the hospital after finishing the measurements and asked to complete a headache diary every hour until 12 h to discharge. The diary included headache characteristics and accompanying symptoms according to the International Headache Society (19), any rescue medication and adverse events. Subjects were allowed to take over the counter rescue medication of their choice at any time.
Headache intensity
Headache intensity was recorded on a verbal rating scale (VRS) from 0 to 10 [0, no headache; 1, a very mild headache (including a feeling of pressing or throbbing); 10, worst imaginable headache] (20).
Cerebral haemodynamics
Middle cerebral artery blood flow velocity
VMCA was recorded bilaterally with transcranial Doppler (TCD) with hand-held 2-MHz probes (Multidop X; DWL, Sippelingen, Germany). Fixed probes were avoided, because they may cause discomfort and even headache (21). Four recordings were taken and averaged at each time point. One recording was a time-averaged mean over 4 s or approximately four cardiac cycles. Identification of the MCA was done as previously described (22). Every TCD recording was performed by the same trained physician (H.W.S.). End tidal CO2 was recorded simultaneously with TCD recordings using an open mask that caused no respiratory resistance (ProPaq Encore®; Welch Allyn Protocol, Beaverton, OR, USA). The measurements at T
20 and T
60 were performed just before and after measurement of regional cerebral blood flow (rCBF) to obtain corresponding values. Flow in the territory of MCA (rCBFMCA) is proportional to the product of VMCA and diameter of the MCA. Assuming that no changes in CBF occur during infusion, the per cent changes in diameter (Δd) can therefore be calculated (23) as:

Diameter of the superficial temporal and radial artery
Diameter of the frontal branch of the STA and RA was measured by high-resolution ultrasonography (Dermascan C; Cortex Technology, Hadsund, Denmark: 20 MHz, bandwidth 15 MHz), as previously described (24, 25).
Cerebral blood flow
Six subjects were randomly allocated to SPECT measurements. The examination was performed with the subject in the supine position in quiet surroundings. Four markers were drawn on the skin to ensure accurate positioning in each acquisition. PetCO2 was measured during each examination (Datex Normocap 200, Roedovre, Denmark). CBF was measured with 133Xe inhalation (Hevesy Laboratory, Ris⊘ National Laboratory, Denmark) and SPECT, using a brain-dedicated gamma camera (Ceraspect; DSI, Waltham, MA, USA). The system uses a stationary annular NaI crystal and a fast rotating collimator. Flow was calculated in each pixel based on the clearance curve, output was the ki value (26). To obtain CBF values, a partition coefficient (λ) of 0.85 was used. Calculation of flow in the perfusion territories of the major cerebral arteries was performed by fitting standard vascular regions of interest (ROI) on transactional slices of the brain. The rCBFMCA was calculated as mean of the right and left side, since there was no statistical difference between the sides.
Vital signs
HR and BP were measured every 10 min using an auto-inflatable cuff (ProPac Encore®; Welch Allyn Protocol). ECG (Cardiofax V; Nihon-Koden, Shinju-ku, Tokyo, Japan) was monitored on a LCD screen and recorded on paper every 10 min.
Data analysis and statistics
All values are presented as mean ±
Calculation of sample size was based on the detection of a difference between carbachol and placebo in the VMCA during the study at 5% significance with 90% power. We assumed that the analyses of variables would show 10% intraindividual variation. A 10% difference between two experimental conditions was taken to be clinically significant. We estimated that 11 subjects should be included for within-person study (27).
The primary end-points were difference in area under the curve (AUC) for headache score (AUCheadache score), VMCA (AUCVMCA), STA (AUCSTA) and RA (AUCRA) between groups during infusion (0–30 min) and postinfusion (30–90 min) phases. The secondary end-points were difference in AUCheart rate, AUCmean arterial pressure, AUCsystolic, AUCdiastolic. Changes in PetCO2 was also evaluated, because VMCA should be corrected with e0.034 for each mmHg change in PetCO2(28).
AUC was calculated according to the trapezium rule (29) to obtain a summary measure and to analyse the differences in response [VMCA, headache score, diameter of RA and STA, mean arterial blood pressure (MAP), HR and PetCO2] between carbachol and placebo. Baseline was subtracted before calculating AUC to reduce variation between sessions within subject. Analysis was performed with a paired two-way t-test, except headache scores, where data are presented as medians and tested with Wilcoxon signed rank test. Baseline was defined as T 0 before the start of infusion of each dose. We tested for period and carry-over effects for all baseline variables with Mann–Whitney test and independent t-test. Adverse event were tested with McNemar test.
For CBF and corresponding PetCO2 values obtained by SPECT and MCA values obtained by transcranial Doppler, continuous variables were analysed for changes over time for each dose separately with univariate analysis of variance with the fixed factors volunteer and time. If overall differences were found, Dunnett's test was applied to characterize which time points were different from baseline. In addition,
All analysis was performed with
Results
Carbachol experimental model of headache
Eleven subjects completed the study on both study days. Five subjects completed CBF measurements on both study days. One subject declined to participate on the second study day (placebo day), but completed the carbachol day with CBF measurement except for completing the headache diary from 2 to 12 h postinfusion due to unspecified reasons. There were missing data on RA values on placebo day in one subject.
There were no differences at baseline for any variables (Table 1). There was no carry-over or period effect for baseline values of headache, MCA, STA, RA, STA, global CBF, regional CBFMCA, MAP, HR or PetCO2.
Baseline values (±

P-value: paired t-test.
Headache
During the immediate phase (0–30 min), six subjects reported headache on carbachol and none on placebo day (Fig. 1). On carbachol day, two of these subjects reported a migraine-like headache (Table 2). The AUC0–30min after carbachol was significantly larger than after placebo (P = 0.042).
Clinical characteristics of nine healthy subjects, who developed headache

A, carbachol day 0–90; b, carbachol day 2–12 h; c, placebo day 0–90 min; d, placebo day 2–12 h; aggr., aggravation of headache by movement; photo, photophobia; phono, phonophobia; heat, heat sensation; migraine like, migraine-like headache.
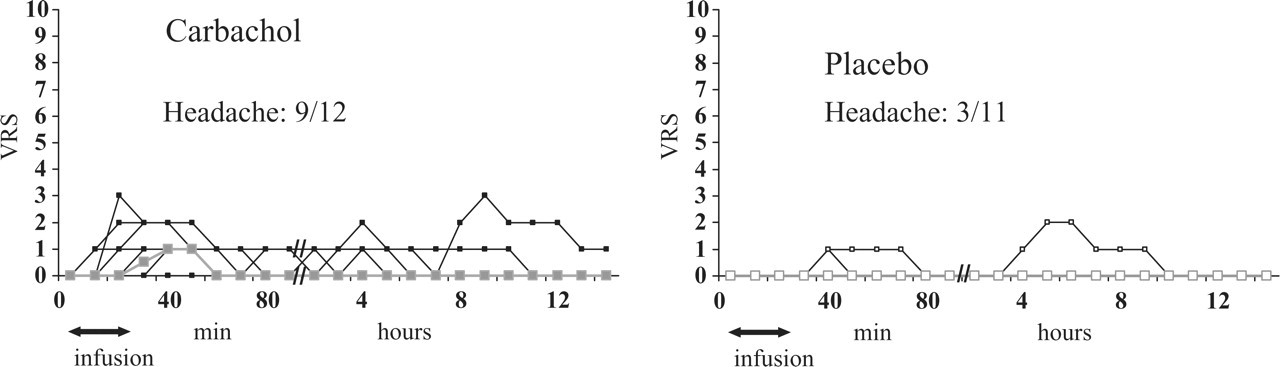
Individual and median headache scores on a verbal rating scale (VRS) from time 0–90 and 2–12 h. There were significantly higher headache responses after carbachol (▪) compared with placebo (□) both during the immediate phase (0–30 min, P = 0.042) and postinfusion phase (30–90 min, P = 0.027). During the postininfusion phase the median headache score peaked (1) 40–50 min after start of infusion on carbachol day. Thick lines show median headache scores.
During the postinfusion phase (30–90 min), seven subjects reported headache on carbachol day and two on placebo day (Figs 1 and 2). The AUC30–90min after carbachol was larger than after placebo (P = 0.027). Median peak headache intensity (1.0) occurred at 40–50 min after start of infusion (Fig. 1). Delayed headache (from 2 to 12 h after infusion) was reported by five subjects on carbachol (Fig. 1 and Table 2) and two of these subjects did not experience headache during the in-hospital phase (0–90). One subject reported delayed headache on placebo, but did not experience headache from 0 to 90 min after placebo infusion. We found no difference in the AUCdelayed headache between the two conditions (P = 0.225). In total, nine subjects developed headache after carbachol compared with three after placebo (McNemar test, P = 0.063).
Middle cerebral artery and regional cerebral blood flow
During the immediate phase (AUC0–30 min), VMCA decreased on carbachol compared with placebo (P = 0.003) (Fig. 3). The mean peak decrease in VMCA compared with baseline occurred 20 min after the start of infusion and was 8.8 ± 7.3% on carbachol and 3.8 ± 4.2% on placebo (Figs 2 and 3). The mean difference in response between carbachol and placebo at T
20 was −5.5% [95% confidence interval (CI) −10.7, −0.2; P = 0.042] (Fig. 3). There was also a difference in the postinfusion phase (AUC30–90min) in carbachol compared with placebo (P = 0.029), due to a small drop in MCA velocity on placebo, where

Individual and mean flow velocities in the middle cerebral arteries (VMCA) assessed by transcranial Doppler ultrasonography. Infusion of carbachol (▪) resulted in a significant decrease in VMCA during the immediate phase (0–30 min) compared with placebo (□) (P = 0.003). Thick lines show mean values.
Given that CBF remained unchanged during carbachol and placebo infusion, the change in MCA diameter was calculated in six subjects. The change in MCA diameter was calculated to be 5.2 ± 4.6% between baseline and T 20 on carbachol and 2.0 ± 2.3% on placebo. The difference between carbachol and placebo was not statistically significant (95% CI −0.4, 6.5; P = 0.052).
There were no differences in the PetCO2 recordings during TCD scans between carbachol and placebo, during either the immediate (AUC0–30min, P = 0.686) or postinfusion phases (AUC30–90min, P = 0.172).
Superficial temporal artery
There was an increase in STA diameter on carbachol compared with placebo during the immediate (AUC0–30min, P = 0.006) and postinfusion phases (AUC30–90min, P = 0.020) (Fig. 4).

Individual and mean diameter of the superficial temporal artery (STA) assessed by high-resolution ultrasonography. During the infusion (P = 0.006) and postinfusion (P = 0.020) phase STA increased significantly after carbachol (▪) compared with placebo (□). Thick lines show mean values.
Mean peak increase in STA diameter compared with baseline occurred 10 min after the start of infusion and was 18.8 ± 15.6% on carbachol and 4.2 ± 9.4% on placebo (Fig. 2). The mean difference in response between carbachol and placebo at T 10 was 14.6% (95% CI −0.4, 29.6; P = 0.055) (Fig. 3), and at T 20 15.7% (95% CI 6.8, 24.6; P = 0.003).

Mean per cent changes from baseline for flow velocity in the middle cerebral arteries (VMCA) (▪), superficial temporal arteries (STA) (□), heart rate (HR) (Δ) and median headache score (•) during the immediate (0–30 min) and postinfusion (30–90 min) phase on carbachol day. VMCA decreased (P = 0.042), while HR (P < 0.001) and STA diameter (P = 0.003) both increased at 20 min from the start of infusion compared with placebo. The median headache score peaked at 40–50 min in the postinfusion phase.
Radial artery
There was no difference in RA diameter on carbachol compared with placebo during the immediate (AUC0–30min, P = 0.200) or postinfusion phases (AUC30–90min, P = 0.203) (Fig. 5).

Individual and mean diameter of the radial artery (RA) assessed by high-resolution ultrasonography. During the infusion (P = 0.200) and postinfusion (P = 0.203) phase RA did not change after carbachol (▪) compared with placebo (□). Thick lines show mean values.
Heart rate and mean arterial blood pressure
We found a significant increase in HR on carbachol compared with placebo during the immediate phase (AUC0–30min, P < 0.001). The mean peak increase in HR compared with baseline occurred 20 min after infusion and was 24.4 ± 9.4% on carbachol and 0.8 ± 3.5% on placebo day (Fig. 2). The mean difference in response between carbachol and placebo at T
20 was 22.6% (95% CI 16.2, 29.0; P < 0.001). We found no difference in HR between carbachol and placebo during the postinfusion phase (AUC30–90min, P = 0.541).
No difference in MAP was found between carbachol and placebo during the immediate (AUC0–30min, P = 0.156) or postinfusion phase (AUC30–90min, P = 0.702). No difference in systolic BP was found between carbachol and placebo during the immediate (AUC0–30min, P = 0.506) or postinfusion phase (AUC30–90min, P = 0.413). No difference in diastolic BP was found between carbachol and placebo during the immediate (AUC0–30min, P = 0.097) or postinfusion phase (AUC30–90min, P = 0.973). However, when exploring changes over time with
Adverse events
Adverse events were recorded and reported during the immediate and postinfusion phases. Increased saliva (P = 0.008), lacrimation (P = 0.031), heat sensation (P = 0.008) and sweating (P = 0.031) were more often reported on carbachol than on placebo (Table 3).
Adverse events recorded and reported in 12 healthy subjects.

Groups compared and tested with McNemar test.
∗ P-values < 0.05.
Discussion
The major outcome of this study is that carbachol induced headache. Furthermore, carbachol also decreased the velocity of the MCA and dilated the STA, whereas no dilation was observed at the RA. In the following we discuss possible mechanisms for the induction of headache and vasodilation.
Mechanisms of vasodilation
Cerebral vasodilation by acetylcholine is dependent on intact endothelial cells (30) and can be inhibited by NOS inhibitors (13). It is probably mediated via muscarinic type 3 receptors (31). This can lead to activation of intracellular endothelial nitric oxide synthase (eNOS) and increased production of NO in endothelial cells (13). NO then diffuses into smooth muscle cells, activates guanylyl cyclase and increases cyclic guanosine monophosphate, which leads to vasodilation by decreasing intracellular calcium (32). Intravenous infusion of the NO donor glyceryl trinitrate (GTN) has been shown to elicit headache in several studies (14, 20, 33, 34). The GTN-induced headache may be due to vasodilation (35) and sensitization of sensory afferent fibres (36). Since GTN easily passes the blood–brain barrier, it has also been suggested that GTN-induced headache could be due to liberation of NO in the CNS, e.g. in the second-order neurons in the nucleus trigeminal caudalis (37, 38). Given that carbachol does not pass the blood–brain barrier, the present results suggest for the first time in man that headache can be induced by activation of endothelial NO production. The intensity of headache found in this study was comparable to the headache caused by GTN in 16 healthy subjects (39). In that study peak median headache was 1.5 compared with 1.0 in the present study and, like the present study, no peak median headache occurred in the delayed phase (2–12 h after infusion).
Vasodilation induced by acetylcholine can also be mediated via production of prostaglandins (40, 41) and endothelium-derived hyperpolarizing factor (40, 42, 43). The contribution of these mediators in dilating vessels seems to vary between vessel sizes (41, 44) and remains to be fully determined. Although carbachol induces changes by affinity of the same receptors (45), a direct comparison between carbachol and acetylcholine is difficult. Carbachol is resistant to degradation by acetylcholine esterase, and it has been shown that the effects of carbachol can be less NO dependent (46). However, it is practically impossible to administer acetylcholine in a human headache model, and carbachol is the best agonist to mimic the effects of acetylcholine.
Haemodynamic effects
Another interesting finding in the present study was that carbachol decreased MCA velocity and dilated the STA, whereas no responsiveness was found for the extracephalic RA. This is in contrast to in vitro studies, where carbachol has been shown to dilate RA (44, 47–49) and in part dependent on endothelial NO production (44). GTN dilates RA by 35% and STA by 41% in healthy subjects (50). The temporal artery dilated 18.8 % in our study, and this was highly significant. The variability by measuring RA is usually smaller than by measuring STA. Therefore, the lack of response of the RA is highly unlikely to be due to a type 2 error. The reason for the lack of dilation of RA after carbachol is unknown, but this heterogeneity in response has previously been shown in intra- vs. extracranial arteries in monkeys (51).
During our experiment, a 24.1% increase in HR was detected 20 min after carbachol infusion. Carbachol is known to be negatively chronotropic (52). By analysis of variance over time it was shown that a drop in MAP and diastolic BP occurred 10 min after carbachol infusion, which might have caused a reflex increase in HR. The increase in HR could also result from carbachol activation of muscarinic type 1 receptors on sympathetic ganglia, which has been proposed as an explanation for atropine-induced bradycardia (53).
Adverse events
We found that increased saliva, lacrimation, heat sensation and sweating were more often reported on carbachol than on placebo. These symptoms are all cholinergic symptoms and indicate that cholinergic receptors were activated. The adverse events on carbachol day may have compromised blindness. However, the adverse events were caused by the physiological response to carbachol and could not be avoided. The present double-blind approach is the best possible way of coping with methodological error.
Parasympathetic neurotransmitters and headache by vasodilation
We have previously investigated other parasympathetic neurotransmitters. Hansen et al. (10) have reported that infusion of VIP induces 8.4% dilation (corrected for CBF) of MCA (vs. 5.2% for carbachol) and 27.8% dilation of STA (vs. 18.8% for carbachol) during infusion. Immediate headache was reported in three of 12 subjects in the postinfusion phase after VIP compared with seven of 12 subjects after carbachol. Thus, despite of the ability of VIP to induce the same or even stronger arterial response as carbachol, it appears that the headache reported in the VIP study was similar to or even less pronounced than headache after carbachol. Birk et al. (9) tested PACAP38, another parasympathetic neurotransmitter, in healthy subjects. Infusion of PACAP38 induced 8.5% dilation of MCA very similar to VIP, but a much more frequent headache (10 out of 11 subjects) compared with both VIP and carbachol. Experimental studies have shown that dilation of cephalic arteries can cause a throbbing headache (54), but the present data on parasympathetic mediators show that headache is not directly linked to the timing and degree of arterial dilation.
In the present study we found a statistical difference in headache response between carbachol and placebo during both the immediate and postinfusion phases, and the median headache peaked 15 min after the end of infusion. The peak of the median headache was preceded by maximal vascular responses, which occurred during the immediate phase of carbachol infusion. Interestingly, peak headache and vasodilation occurred simultaneously in GTN studies (20, 33). This may indicate that the mechanisms of GTN- and carbachol-induced headaches differ, and in the following section we discuss other possible mechanisms.
Effect of carbachol on sensory activation and mast cell degranulation
Dural vessels are pain sensitive and have been implicated in headache pathogenesis. Nerve fibres containing acetylcholine-producing enzymes are found in cerebral (6, 11), dural (5) and temporal (12) arteries in close relation to the advential-medial borders of the vessel wall. In the rat dura, muscarinic (55) as well as nicotinic (56–58) receptors are found on trigeminal ganglion neurons. Furthermore, it has been shown that dural sensory nerve fibres lie in close apposition to mast cells as well as parasympathetic nerve fibres (59) in the rat. Mast cells are also found in the human cerebrum (11) and dura mater (59) adjacent to blood vessels.
Animal studies have shown that acetylcholine can activate nociceptive C-fibres in the cornea (17) and skin (15). Carbachol is also a very effective inducer of mast cell degranulation (60–62), including dural mast cells (59). Intradermal injections of acetylcholine induce pain and activation of nociceptive C-fibres in humans (63). When applied to a blister base (64) or as intra-arterial injection (65), acetylcholine causes localized pain in humans.
Carbachol-induced headache may be due to direct activation of sensory neurons or to mast cell degranulation. Delepine et al. (66) have shown that intra-arterial infusion of carbachol in doses similar to the present study induce plasma protein extravasation (PPE) in the rat dura mater, which is blocked by a muscarine receptor antagonist. The authors suggested that the development of PPE could be a sign of direct activation of sensory C-fibres and mast cell degranulation, because trigeminal sensory fibre stimulation induces PPE as well as mast cell activation (67, 68). Furthermore, Levy et al. (69) have shown that mast cell degranulation led to neuronal excitation in afferent nociceptive C-fibres innervating the dura mater and activation of second-order brainstem neurons. Neurotransmitters released from activated sensory fibres may also activate mast cells (67). The result is a vicious circle, because the activation of mast cells releases substances that sensitize sensory fibres. Collectively, these data suggest that carbachol can degranulate dural mast cells and sensitize sensory afferent fibres in the dura, which could be the mechanism of carbachol-induced headache.
Conclusion
Carbachol induced headache and cephalic vasodilation in healthy subjects. Possible mechanisms for the induction may be activation of endothelial muscarinic receptors in the central arteries stimulating NO production. Alternatively, carbachol may be causing headache by degranulating mast cells and activation of nociceptive fibres in the dura and/or extracranial tissue. The effect of carbachol on headache patients and further exploration of the mechanisms involved should be the subject of future studies.
Footnotes
Acknowledgements
The authors thank lab technicians Kirsten Brunsgaard, Lene Elkjær and Annette Foldager for their dedicated and excellent assistance. Jens Rokkedal, MD, MDSci and J⊘rgen Jeppesen, MD, MDSci from the Cardiology Department and Internal Medicine at Glostrup Hospital are acknowledged for their advice on cardiovascular safety parameters. We also thank Peter Dalgaard from the Department of Biostatistics, University of Copenhagen for statistical advice on the study. The study was supported by the Lundbeck Foundation via the Lundbeck Foundation Centre for Neurovascular Signalling (LUCENS), the Cool Sorption Foundation and the Mauritzen La Fontaine Foundation.