
Abstract
Introduction
Carbon monoxide (CO) is an endogenously produced signalling molecule that has a role in nociceptive processing and cerebral vasodilatation. We hypothesized that inhalation of CO would induce headache and vasodilation of cephalic and extracephalic arteries.
Methods
In a randomized, double-blind, placebo-controlled crossover design, 12 healthy volunteers were allocated to inhalation of CO (carboxyhemoglobin 22%) or placebo on two separate days. Headache was scored on a verbal rating scale from 0–10. We recorded mean blood velocity in the middle cerebral artery (VMCA) by transcranial Doppler, diameter of the superficial temporal artery (STA) and radial artery (RA) by high-resolution ultrasonography and facial skin blood flow by laser speckle contrast imaging.
Results
Ten volunteers developed headache after CO compared to six after placebo. The area under the curve for headache (0–12 hours) was increased after CO compared with placebo (p = 0.021). CO increased VMCA (p = 0.002) and facial skin blood flow (p = 0.012), but did not change the diameter of the STA (p = 0.060) and RA (p = 0.433).
Conclusion
In conclusion, the study demonstrated that CO caused mild prolonged headache but no arterial dilatation in healthy volunteers. We suggest this may be caused by a combination of hypoxic and direct cellular effects of CO.
Introduction
Carbon monoxide (CO) is a gaseous signalling molecule produced endogenously in the human body by the enzyme haeme oxygenase (HO) (1). CO is involved in multiple biological processes, including neurotransmission (2) and vasodilatation (3–7). One of the initial symptoms of acute CO poisoning is headache (8,9). Headache is also reported at low non-toxic CO levels (8,10,11). The molecular mechanism behind the headache inducing effect of CO is not fully clarified (12). CO may be involved in pain signalling via cyclic guanosine monophosphate (cGMP) pathways (1,2,13) and via interactions with nitric oxide (for review, see Arngrim et al. (12)), which plays an important role in primary headaches (14). Human exposure to CO inhalation with a carboxyhemoglobin concentration of 20% increases the cerebral blood flow by 26% (3). Ex vivo experiments have suggested a dilatory effect of CO on cerebral and systemic arteries (4–7). Taken together, these data suggest that CO may be an important molecule in primary headaches (12). To date, there has been no systematic investigation of the headache inducing effect of CO in healthy volunteers. We hypothesized that CO would induce headache and dilate cranial arteries. To test this hypothesis, we conducted a double-blind placebo controlled crossover study in healthy volunteers.
Material and methods
We first conducted a pilot experiment to determine the optimal dose of CO that induced headache and the optimal method to measure carboxyhemoglobin (COHb). We then examined the headache inducing and hemodynamic effect of a fixed dose of CO in healthy volunteers.
We recruited healthy volunteers via an announcement on a Danish website for recruitment of volunteers for health research (forsoegsperson.dk). Exclusion criteria were: A history of migraine or any other type of headache (except episodic tension type headache for less than five days per month); first-degree relatives with migraine; any daily medication apart from oral contraceptives; any somatic or psychiatric disease; any headache 48 h before the start of each experiment, intake of coffee, tea, cocoa, alcohol, tobacco or other methylxanthine-containing foods or beverages 12 h prior to the study days. A full medical history and examination was performed prior to inclusion.
The study was approved by the Ethics Committee of the Capital Region of Denmark (H-3-2013-194) and the Danish Data Protection Agency. The study was registered at Clinicaltrials.gov (ID: NCT02066558). All participants provided their written informed consent to participate in the study after detailed oral and written information, in accordance with the Declaration of Helsinki 2013 version.
Design of the pilot study
In a double-blinded two-way crossover design, three healthy volunteers were randomly allocated to inhalation of two doses of CO (COHb 10% and 22%) on two separate days with a minimum interval of one week (Figure 1(a)). In the pilot study, COHb was measured every 10 min from 0 to 60 min and every 15 min from 60 to 180 min after the first CO inhalation by three methods: In capillary blood samples (both study days), arterial blood samples from a catheter in the radial artery (one study day), and by COHb oximeter (Masimo, Hannover, Germany) (both study days). The purpose was to determine the optimal dose of CO in the main experiment that induced headache and to choose a reliable method for COHb measurement. Besides the COHb level, measuring methods and study duration (180 min), all methods used in the pilot were the same as in the main experiment. The severity and duration of CO exposure in this study was based on earlier studies, and entails no risk in healthy subjects (15,16).
(a) Study flow chart; (b) Design of main experiment.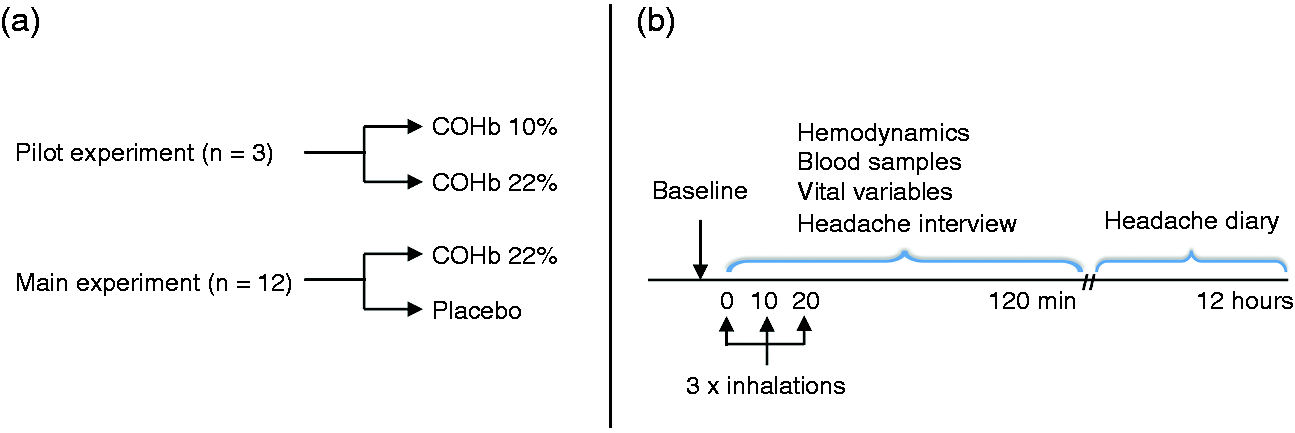
Design of the main experiment
In a double-blind placebo-controlled crossover design, the participants were randomly allocated to inhalation of CO (COHb 22%) or placebo (atmospheric air) through a mask on two separate days, with a minimum interval of one week (Figure 1(a)). The COHb concentration in the main experiment was based on the pilot experiment.
All participants arrived headache-free at the laboratory at 08:00 on each study day. All measurements were performed by the same skilled technicians (LE and WG) in a quiet room with the room temperature between 23 and 25℃. The participants were placed in a supine position and the VMCA, diameter of superficial temporal artery and radial artery were measured before inhalation (baseline) and every 10 min from 30 to 120 min. Headache intensity, blood flow in the facial microvasculature and vital signs were monitored with regard to electrocardiogram (ECG) (Cardiofax V, Nihon-Cohden, Shinjuku-ku, Tokyo, Japan), mean arterial blood pressure (MAP), heart rate (HR), respiration frequency (RF) and end-tidal CO2 (PETCO2) (ProPaq Encore®; Beaverton, OR, USA) at baseline and then every 10 min until 120 min had passed. Two capillary blood samples were collected at baseline and every 10 min from 0 to 30 min, and every 30 min from 30 to 120 min. Blood samples were analysed immediately after sampling to determine concentration of COHb, haemoglobin (ctHb), potassium, sodium and glucose (blood gas analyser, Radiometer, Copenhagen, Denmark). Apart from analysis of blood samples, all measurements were performed by the blinded technicians (LE and WG). After finishing the measurements, the participants were discharged from the hospital and asked to complete a headache diary every hour until 12 h after the first inhalation (Figure 1(b)).
Carbon monoxide
The participants were exposed to CO through a 2 m tube, a one-way valve and a tight fitting full-face mask (Hans Rudolph). The inhalation lasted approximately three minutes and was performed three times: At baseline (time 0), and after the measurements at 10 and 20 min. During the first inhalation, 200 ml of CO (99.997% purity, Strandmoellen, Denmark) was added to the tube by four 50 ml syringes. COHb from capillary blood samples was determined and the volume of CO (ml) necessary to increase COHb by 1 mM was calculated (200 ml CO/(COHb after inhalation – baseline COHb)). Based on this value and the COHb concentration, the volume of CO needed for the second and third inhalation to reach COHb 22% was calculated. The participants were instructed to take slow deep breaths during the inhalations. The inhalation procedure was performed by a blinded technician (LE)/investigator (JB), and an unblinded physician (NA) according to a randomization scheme.
Headache and adverse events
We told the participants that we were investigating the possible headache inducing effects of CO. The timing, occurrence or character of a possible headache was not specified. Headache intensity was recorded on a numerical rating scale (NRS) from 0–10 (0: no headache; 1: a very mild headache (including a feeling of pressing or throbbing pre-pain); 10: worst imaginable headache) (17). We also recorded headache characteristics (intensity [moderate to severe headache intensity was considered ≥ 4 on NRS], quality, location, aggravation by physical activity), and associated symptoms (nausea, photophobia and phonophobia). Two laboratory technicians (LE and WG) extracted and double-checked headache data blinded.
Mean blood velocity of middle cerebral artery (VMCA)
Mean VMCA was recorded bilaterally using transcranial Doppler (TCD; Doppler BoxX, DWL, Singen, Germany) with hand-held 2-MHz probes as previously described (18,19). Mean VMCA was acquired as time-averages over four cardiac cycles calculated using in-house developed Matlab scripts (Mathworks, Natick, MA, USA). At each time point, four consecutive recordings of mean VMCA were acquired and averaged for the further analysis.
Diameter of superficial arteries
The diameter of the frontal branch of the superficial temporal artery (STA) and the radial artery (RA) was measured using high-resolution ultrasonography, 20 MHz, bandwidth 15 MHz (Dermascan C, Hadsund, Denmark) as previously described (20,21).
Facial skin blood flow
Facial skin blood flow was measured by laser speckle contrast imager, moorFLPI (Moor Instruments, Devon, UK). The instrument head with the camera was positioned 30 cm straight above the face of the participants and focus was optimized. Calibration of the imager was done using a reference flux signal generated by the laser light scattered from a suspension of polystyrene microspheres in water undergoing thermal or Brownian motion. We made measurements of the entire face as previously described (22,23). The participants were instructed to lie completely still with eyes closed during measurements. The recorded contrast images were processed to produce a scaled color-coded live flux image (red equaled high perfusion, blue equaled low perfusion), which correlated with the blood flow velocity in the face skin. The mean perfusion of the entire face was calculated at each time point based on the flux images.
Data analysis and statistics
Data are presented as means ± standard deviation (SD), except headache scores, which are presented as median and individual values. Percentage changes are presented with a 95% confidence interval (CI). Baseline was defined as t0 before the start of test gas inhalation.
Sample size was calculated based on detection of a difference between CO and placebo in incidence of headache at a 5% significance (two-tailed) level and 90% power. Based on the anticipation that CO would induce headache in 75% of the participants versus 10% after placebo, we estimated that 12 participants should be included.
The primary endpoints of the main experiment were the difference between CO and placebo for the following data: (i) 0 to 12 h: Incidence of headache and area under the curve (AUC) for headache score; (ii) 0 to 120 min: AUC of mean VMCA; diameter of STA, diameter of RA and facial skin blood flow. The secondary end-points were the difference in AUC from 0 to 120 min of HR, MAP, RF and PetCO2, capillary blood samples (COHb, hemoglobin, sodium, potassium and glucose), and the incidence of adverse events between CO and placebo. The incidence of headache and adverse events was analysed as binary categorical data with McNemar’s test. We calculated AUC according to the trapezium rule to obtain a summary measure and to analyse the difference in response between CO and placebo (24). The baseline was subtracted before calculating AUC to reduce within-participant variation between sessions. AUC and baseline differences of all variables between CO and placebo were compared using the non-parametric Wilcoxon signed rank test. We tested for period and carry-over effects for all baseline variables with the Mann-Whitney test. All statistical analyses were performed with SPSS version 23.0 (Chicago, IL, USA). We made no adjustment for multiple analyses. Five percent (p < 0.05) was accepted as the level of significance.
Results
Pilot experiment
Three healthy volunteers (males, age 24, 24 and 25 years) completed both study days. All healthy volunteers reported headache on both study days. The headache scores were numerically higher and of longer duration after COHb 22% compared to COHb 10% (Figure 2(a)). COHb measured in arterial and capillary blood samples were equal, but different from the values measured by the COHb oximeter (Figure 2(b)).
Pilot experiment. (a) Individual headache intensity after carbon monoxide (CO) (Carboxyhemoglobin (COHb) 10% and 22%) in three healthy volunteers (participant 1 in blue, participant 2 in red, participant 3 in green). (b) COHb measured in arterial and capillary blood samples and by COHb oximeter on three study days (participant 1 in blue, participant 2 in red, participant 3 in green).
Main experiment
Twelve healthy volunteers (nine females, mean age 26 years, range 24–34 years) completed both study days. There were no differences in baseline values between the two study days apart from a small difference in diameter of STA (CO: 0.89 ± 0.25 mm, placebo: 0.97 ±0.25 mm, p = 0.033) and MAP (CO: 74.13 ±4.66 mmHg, placebo: 77.00 ± 4.68 mmHg, p = 0.003). There was no carry-over or period effect for baseline values except for a period effect of MAP (day1–day2, p = 0.018).
Headache
Characteristics of peak headache after CO and placebo inhalation in healthy volunteers (0 to 12 h).

Localization/intensity/quality/aggravation by movement.
Photophobia/phonophobia/nausea.
Pain relief 50% within 2 h.
Participant 7 had headache that fulfilled the migraine-like criteria at 20 min (left side, intensity 1, throbbing and pressing, aggravation by movement, photo- and phonophobia, and at 12 h (bilateral, intensity 1, throbbing, aggravation by movement and nausea.
ND: not determined; h: hours; min: minutes; bilat: bilateral.

Headache intensity and carboxyhemoglobin levels. Individual and median headache intensity shown on the right y axis after carbon monoxide (CO) (a) and placebo (b) in 12 healthy volunteers. Mean carboxyhemoglobin (COHb) concentration (95% CI) is shown on the left y-axis. The time course of decrease in COHb after 2 h has been extrapolated to 0% (a). The median peak headache score after CO was 1 (range 0–4). The median time to peak headache after CO inhalation was 3.5 h. The median duration of headache after CO inhalation was 3.5 h (range 20 min – 11 h).
Hemodynamics
Mean VMCA increased after CO compared to placebo (AUC0–120 min, p = 0.002) (Figure 4). The mean peak increase in VMCA after CO was 31% (range 16–40%). VMCA peaked at 40 min after CO inhalation (mean increase 25% (range 9–40%).
Hemodynamic changes after CO (a) and placebo (b). Left y-axis: Mean percentual (95% CI) hemodynamic and heart rate changes from baseline. Right y axis: mean % carboxyhemglobin level (95% CI).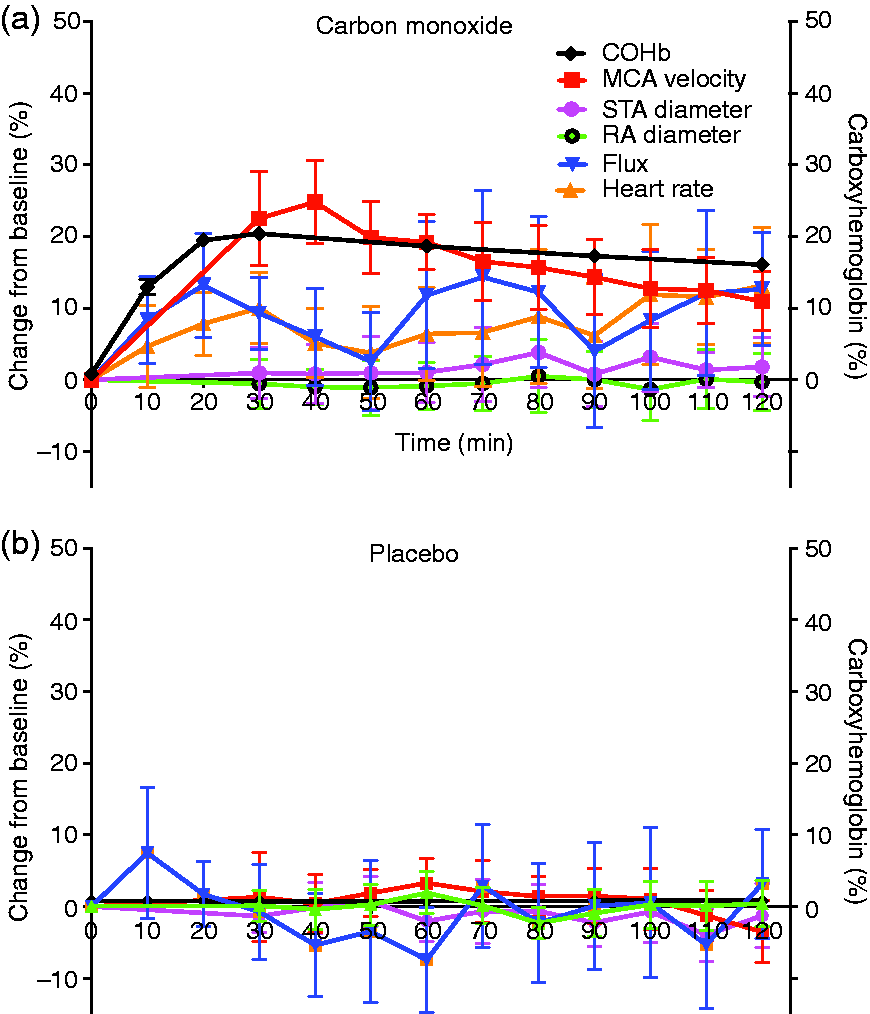
There were no differences in AUC0–120min of STA diameter (p = 0.060) or RA diameter (p = 0.433) after CO compared to placebo (Figure 4).
The facial skin blood flow increased after CO compared to placebo (AUC0–120 min, p = 0.012) (Figure 4).
Blood samples
After CO inhalation, the healthy volunteers reached a mean COHb of 20.4% (SD = 0.8%) (Figure 3(a)) after inhalation of a mean CO volume of 368 ml (SD =47 ml). Extrapolation of COHb washout showed that the participants reached baseline COHb approximately 8 h after baseline (Figure 3(a)). AUC0–120 min of COHb was higher (p = 0.002) and there was no difference in AUC0–120 min of haemoglobin (p = 0.875), glucose (p =0.656), potassium (p = 0.308) and sodium (p = 0.099) after CO compared to placebo (Figure 5(a)).
Capillary blood samples and vital variables. Mean ± SD values of capillary blood samples (a) and vital variables (b) 0 to 120 min after inhalation of carbon monoxide and placebo.
Vital variables and adverse events
Incidence of adverse events 0 to 120 min after inhalation of CO or placebo*.
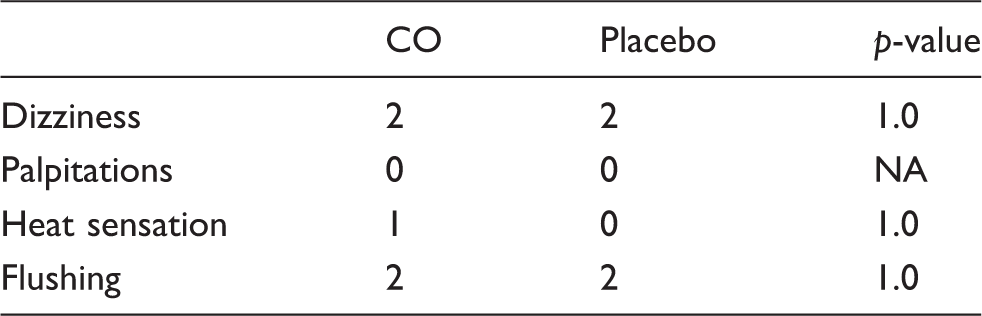
NA: not applicable.
In addition, the following adverse events were reported by a few participants: after CO, tiredness (n = 2), urination urge (n = 2), general/leg numbness (n = 2); after placebo, tiredness (n = 2).
p-value: McNemars test.
Discussion
The major finding of the present study was that CO inhalation in a human headache model provoked mild and prolonged headache in healthy volunteers. Furthermore, CO increased mean VMCA and facial skin blood flow, whereas no changes were observed in the diameter of the superficial temporal artery (STA) and the radial artery (RA).
Present findings in relations to previous CO exposure studies
In the present study, healthy volunteers reported mild bilateral CO-induced headache of pressing character after a mean peak COHb of 21%. This corresponds well with the diagnostic criteria of CO-induced headache in the ICHD-3 (beta), which describes headache caused by CO exposure as bilateral and correlated to the COHb levels (25).
Previous headache data from accidental CO exposures suggested that the severity of CO-induced symptoms, including headache, is not related to the actual COHb concentration (9,26), but more related to the duration of CO exposure (9). In contrast, we showed that very short but intense exposure (max 10 min) to CO is sufficient to induce prolonged headache. Only three earlier studies investigated the clinical effects of pure CO inhalation (11,27,28). In 1922, Sayers et al. performed CO self-exposure studies (27) and reported tightness across the forehead that developed into headache on exertion at COHb 10–20% and headache with worsening on exertion at COHb 20–25% (27). Steward et al. (11) reported no headache after exposure to low CO concentrations for eight to 24 h, reaching COHb in the range of 6–12% in healthy volunteers, but headache after 10 min exposure to high CO levels reaching a COHb of 9% and 12% in two healthy volunteers (28). Both studies reported worsening of CO-induced headache on exertion (11,27). In our study, five healthy volunteers reported worsening of CO-induced headache on exertion. Notably, all previous studies were performed without proper headache registration, blinding and placebo control.
The CO-induced headache was mild, with a median intensity and median peak headache NRS score of 1, which is similar to headache induced by the NO donor glyceryl trinitrate (GTN) in healthy volunteers (median NRS 1.5) (29). The incidence of headache of 83% is similar to earlier studies with GTN (92–100%) (29,30), hypoxia (73%) (31) and cilostazol (92%) (32) in healthy volunteers. The incidence of headache after placebo of 50% is relatively high, but similar incidence has been reported after placebo to GTN (42%) (30). The CO headache intensified 3.5 to 6 h after the last CO inhalation, and the median duration of headache was 3.5 h (range 20 min to 11 h). A similar “delayed” intensification of headache after CO inhalation was reported by Steward et al. (11). In contrast, GTN only provokes an immediate headache during the 20 min infusion in healthy volunteers (17) and a delayed migraine-like headache in migraine patients (33).
In the present study, we only investigated the effect of short, intense exposure to CO. Interestingly, CO levels in smokers may be increased by up to 15%, and smoking may be associated with an increased risk of migraine with aura (34). Furthermore, an association between ambient air pollution (fine particle levels including CO) and daily clinic visits for migraine has been reported (35,36). Thus, chronic exposure to CO may play a role in migraine.
Present hemodynamic findings in relation to previous studies
Paulson et al. (3) showed that inhalation of CO resulting in a mean COHb 20% increased human cerebral blood flow (CBF) by 26% using the intra-arterial xenon clearance method, which is similar to the 31% mean peak increase in VMCA in the present study. In vivo animal studies reported a direct vasodilatory effect of CO on cerebral arterioles (37,38). Arterial dilatory effects have also been suggested based on ex vivo studies of dog basilar artery (4), mouse MCA (5) and human internal thoracic and radial artery grafts (6). To our knowledge, the present study is the first to directly assess the blood velocity in MCA, the diameter of a cranial and peripheral artery and the blood flow in the microvasculature of the face after inhalation of CO. Interestingly, CO did not affect the diameter of the STA and RA, but increased the dermal flow to the face. This suggest that inhalation of CO has no vasodilatory effect on arteries but mainly affects the microvasculature. The discrepancy between our results and the previous data on radial artery grafts (6) may be explained by the different methods applied, especially concentrations of CO. The ex vivo radial artery graft rings were pre-contracted with norepinephrine in an organ chamber, which subsequently was bubbled with CO (100%) until tension stability (max CO concentration 3.10−5 M) (6). In pigs, the dilatory effect of topically applied CO on pial arterioles is dose dependent (37).
Mechanisms behind CO induced hemodynamic changes
Earlier, the CO induced change in CBF was regarded as a hypoxic induced compensatory response. CO has a 250 times higher affinity to heme than oxygen, and also impairs the release of heme bound oxygen (39,40). In the present study, the participants had an oxygen saturation of approximately 79% after the end of CO inhalation. This level of hypoxia increases CBF (41) to a similar extent to that reported during COHb 20% (3). However, 2 h of hypoxia (SpO2 72%) dilated the STA 20% and MCA 10% in healthy volunteers (31). Furthermore, 3 h of hypoxia (SpO2 75%) increased CBF but did not increase VMCA measured using TCD (42). This suggests different hemodynamic effects of CO and pure hypoxia. CO does not only bind haemoglobin, approximately 15% of CO in the body is bound to extravascular heme-proteins (43). The tissue uptake of CO in the brain during CO inhalation has been suggested to be relatively high due the rapid metabolism in the brain causing a low oxygen tension, in combination with a relatively slow saturation of haemoglobin with CO in erythrocytes and a high CO tension in the pulmonary veins (44,45). Thus, free CO may reach the brain and exert direct cellular effects by binding to different heme-proteins and thus have direct effects on the cerebral hemodynamics. This is supported by numerous studies showing arteriolar dilatation after CO exposure in the open cranial window model in rats (37,38). Interestingly, this dilatation can be blocked by inhibitors of calcium-gated-potassium channels (KCa+-channels), soluble guanlylate synclase and nitric oxide synthase inhibitors (For review see Arngrim et al. (12)). Thus, KCa+-channels, cGMP signalling pathways and NO may be involved in the hemodynamic effects of CO.
Mechanisms behind CO induced headache
The headache inducing effects are likely due to a combination of CO-induced hypoxia and the direct cellular effects of CO. Thus, the occurrence and severity of headache caused by CO is likely to depend on both the COHb level (i.e. the severity of hypoxia) and the amount of free CO that reaches the nervous system, which probably will increase with increased CO tension in the inhaled air and with long duration of CO exposure.
Hypoxia (SpO2 72%) induces headache in healthy volunteers and migraine attacks in migraine with aura patients (31). The delayed intensification of headache after CO inhalation is unlikely to be caused solely by hypoxia, since the hypoxia at 4 h (COHb approximately 10%) is very mild, corresponding to an oxygen saturation (SpO2) of approximately 90%.
Abundant studies with manipulation of the endogenous CO production in animal pain model studies suggest that CO is a pain modulating neurotransmitter via activation of cGMP pathways, interactions with NO, oxidative stress, glutamate and regulation of nociception-related gene expression (for review see Arngrim et al. (12)). These mechanisms may also be involved in headache generation. Activation of the cGMP pathway is likely to play a central role in the pathophysiology of migraine and other headaches (14). CO has high affinity to soluble guanylate cyclase (sGC), and binding stimulates the activity of sGC and thus cGMP formation (2,13). Interestingly, CO has many similarities to NO, and NO plays an important role in primary headaches (14). NO is also an endogenously produced gaseous neurotransmitter that acts via the cGMP pathways (14). Both gases bind haemoglobin and inhibit oxidative phosphorylation by binding to cyctochrome c oxidases. The two gases also affect each other’s synthesis. Like NO, CO can be generated from the vascular wall and has vasoactive effects. However, the interaction between CO and NO and its possible implication in headache generation is not fully understood (for review see Arngrim et al. (12)). In contrast to CO, the NO donor GTN does not increase CBF but decreases VMCA and dilates STA, RA and MCA (30,46,47). Interestingly, sumatriptan may prevent GTN induced immediate headache in healthy volunteers (48). CO-induced headache may respond to sumatriptan (49). It would be interesting to further investigate the effect of sumatriptan in the CO headache model.
To date, all headache and migraine provoking substances are vasodilators (50). Furthermore, migraine attacks are associated with modest dilation of intracranial arteries, but no dilation of extracranial arteries (51). Heme oxygenase is highly expressed in large and small cerebral vessels, astrocytes and neurons (37,52,53). Although, our study suggests the vasoactive effects of CO are primarily in the microvasculature, we cannot exclude that CO released from the vascular wall may cause headache by stimulation of perivascular nociceptors (54).
There was a small difference in diameter of the STA and MAP at baseline on the two study days and a period effect for baseline MAP. We cannot explain this, but it is unlikely to affect the results since there was no difference in the exact values or percentage changes from baseline on the two study days.
Conclusion
CO caused prolonged mild headache and increased blood velocity in the MCA, but did not dilate STA and RA in healthy volunteers. We suggest that the headache-inducing properties and hemodynamic effects of CO are caused by a combination of hypoxic and direct cellular effects. The direct cellular mechanisms may include cGMP-pathways and NO. Endogenously, CO may play a role in the pathophysiology of primary headache. The effect of CO inhalation in migraine patients remains to be investigated.
Clinical implications
CO inhalation provoked mild and prolonged headache in healthy volunteers. CO increased blood velocity in the middle cerebral artery. CO did not change the diameter of the superficial temporal artery and the radial artery. We suggest this human headache model can be used to investigate the role of CO in migraine.
Footnotes
Acknowledgement
The authors thank lab technicians Lene Elkjær and Winnie Grønning for expert assistance.
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The study was supported by the Capital Region of Denmark Foundation for Health Research (A4620), the Lundbeck Foundation (R155-2014-171), the Novo Nordic Foundation (NNF11OC1014333), Danish Council for Independent Research (DFF-4004-00169B), Simon Fougner Hartmanns Familiefond, and the European Union’s Seventh Framework programme (2007-2003) under grant agreement no. 602633.