
Abstract
Familial hemiplegic migraine (FHM) is a rare type of migraine with aura. Mutations in three genes have been described in FHM patients: CACNA1A (FHM1), ATP1A2 (FHM2) and SCN1A (FHM3). We screened 27 Spanish patients with hemiplegic migraine (HM), basilar-type migraine or childhood periodic syndromes (CPS) for mutations in these three genes. Two novel CACNA1A variants, p.Val581Met and p.Tyr1245Cys, and a previously annotated change, p.Cys1534Ser, were identified in individuals with HM, although they have not yet been proven to be pathogenic. Interestingly, p.Tyr1245Cys was detected in a patient displaying a changing, age-specific phenotype that began as benign paroxysmal torticollis of infancy, evolving into benign paroxysmal vertigo of childhood and later becoming HM. This is the first instance of a specific non-synonymous base change being described in a subject affected with CPS. The fact that the molecular screen identified non-synonymous changes in< 15± of our HM patients further stresses the genetic heterogeneity underlying the presumably monogenic forms of migraine.
Introduction
Familial hemiplegic migraine (FHM) (MIM# 141599) and sporadic hemiplegic migraine (SHM) are rare types of migraine with aura (MA). The deciphering of the first two molecular defects underlying the condition has led to its being classified into subtypes. In FHM1, missense mutations in the CACNA1A gene on chromosome 19p13.13, encoding the α subunit of the neuronal P/Q-type calcium channel (CACNA1A), were first reported in five unrelated FHM families (1). To date, at least 18 different CACNA1A missense mutations have been reported in FHM families (2). Most of these mutations recur very infrequently, with the exception of p.Thr666Met, found in 20 families worldwide, and p.Arg583Gln, reported in six families (3). About half of FHM families show additional signs of progressive cerebellar ataxia (4) or, less frequently, epileptic seizures (5), cognitive dysfunction (6) or migraine coma (7).
Mutations in the CACNA1A gene also cause episodic ataxia type 2 (EA-2) (MIM#108500) and spinocerebellar ataxia type 6 (SCA6) (MIM#183086). Most of the EA-2-related CACNA1A mutations are nonsense, splice site, small deletions or insertions which disrupt the open reading frame and result in a truncated protein, whereas SCA6 is usually caused by expansion of an unstable CAG repeat on the C-terminal region of the protein (8). However, missense mutations have also been identified in SCA6 and EA-2.
Conversely, FHM2 is linked to mutations in the ATP1A2 gene on chromosome 1q23.2. More than 30 FHM2 missense mutations have been identified in this gene so far (2, 3, 9), but also in other phenotypes including alternating hemiplegia of childhood (10), MA, migraine without aura (MO) (11) and basilar-type migraine (BM) (12).
Recently, a third gene has been associated with FHM. The same mutation, p.Gln1489Lys, has been identified in three independent FHM families, now classified as FHM3, in the SCN1A gene, which encodes the α subunit of the neuronal voltage-gated type I sodium channel (13).
Some clinical syndromes that are precursors of migraine in young patients, including cyclical vomiting (CV), abdominal migraine and benign paroxysmal vertigo in childhood (BPV), have recently been reclassified by the International Headache Society (IHS) as childhood periodic syndromes (CPS). Although these syndromes have been known for decades (14, 15), their suggested relationship to migraine remains a matter of debate, as is the case with another possible subtype of migraine precursor, benign paroxysmal torticollis of infancy (BPT). In addition, there is very little genetic evidence to date linking familial migraine and CPS (16).
Here we report the clinical manifestations of a series of 27 Spanish patients diagnosed with hemiplegic migraine (HM), BM or CPS and the results of a molecular analysis of the three known FHM genes that allowed the identification of three CACNA1A non-synonymous base changes.
Materials and methods
Subjects
This study included 27 unrelated Spanish patients referred to Vall d'Hebron University Hospital, Barcelona, during the period 1999–2004 with diagnoses of HM, BM or CPS, the migraine phenotypes previously associated with mutations in the CACNA1A, ATP1A2 and SCN1A genes. CPS presented as BPV or BPT [the latter being considered as a possible CPS by virtue of its present classification and encoded A1.3.5 in the International Classification of Headache Disorders (ICHD-II) appendix (17)]. Twenty-two patients were directly interviewed and examined by one of the authors (A.M.), and information about the remaining five cases was obtained from referring physicians and completed through telephone interview. Positive family history in first- or second-degree relatives was elicited from the patients or from their parents in case of paediatric patients. In kindreds with positive family history, at least one affected relative was directly interviewed; the diagnosis of FHM was established by the presence of two or more cases of MA including motor weakness. The diagnosis of CPS was based on the presence of recurrent torticollis, vertigo, vomiting and/or behavioural changes in neurologically and audiologically intact children, and was later confirmed in all cases following the guidelines given by the IHS in the ICHD-II (17). The clinical diagnosis of migraine types was initially established in accordance with the ICHD-I criteria (18) and later confirmed according to ICHD-II. For genetic studies 64 unrelated control individuals were recruited from the Blood Extraction Unit of the Vall d'Hebron University Hospital. They were Spanish, Caucasoid and lacked any history of recurrent or disabling headache, as did their first-degree relatives.
Samples
Blood venous samples were obtained from 27 probands and from 64 unrelated non-migraineurs. Genomic DNA was isolated using the QIAamp DNA Blood Maxi Kit (Qiagen, Hilden, Germany) after each subject had provided written informed consent for DNA analysis and the local ethics committee had approved the study, which followed the guidelines of the Helsinki Declaration.
DNA and mutation analysis
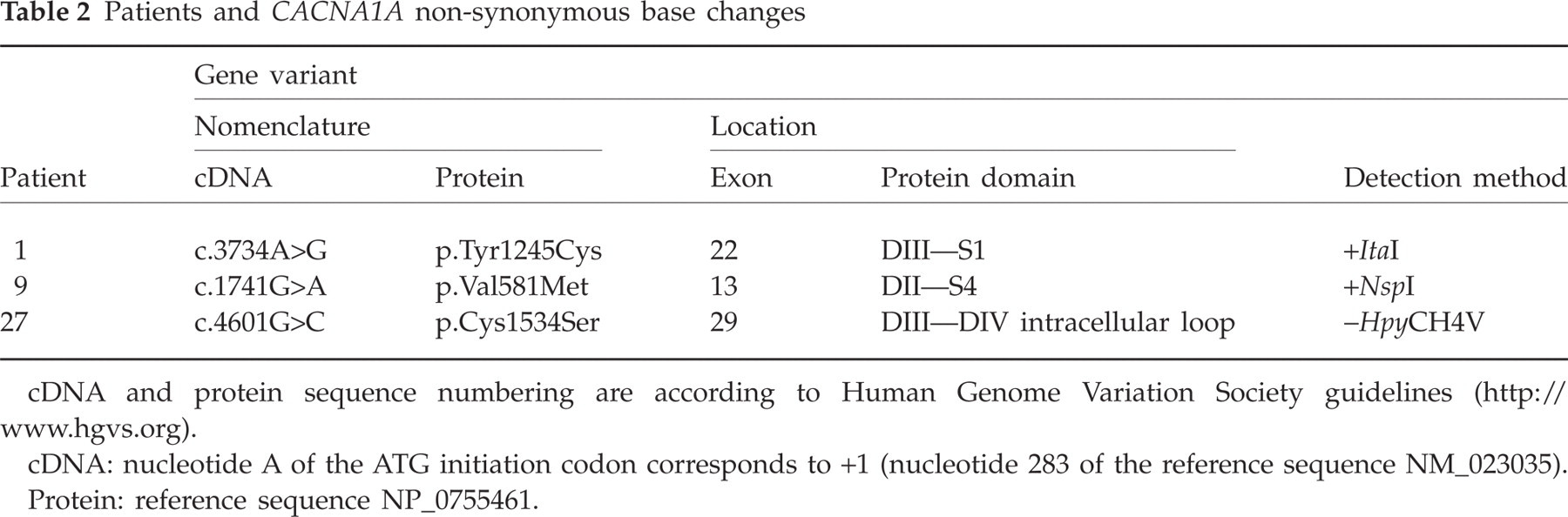
The 47 exons of the CACNA1A gene, the 23 exons of the ATP1A2 gene and exon 23 of the SCN1A gene and their corresponding exon/intron junctions, including splice sites and branch points, were polymerase chain reaction (PCR)-amplified and sequenced in 42, 16 and one independent PCR products, respectively. The primers were designed using Primer3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) (19). Primer sequences are available as supplementary material (Tables S1 and S2). The details of the PCR procedures are available from the authors upon request. The PCR products were purified and sequenced (ABI PRISM 3700 DNA analyser; Applied Biosystems, Foster City, CA, USA). The identified gene variants were confirmed by digestion of the corresponding PCR product with the appropriate restriction enzyme: NspI (p.Val581Met), ItaI (p.Tyr1245Cys) and HpyCH4V (p.Cys1534Ser).
Sixty-four unrelated healthy Spanish individuals were screened for the presence of the gene variants identified in the patients by either restriction enzyme cleavage of PCR products or single-strand conformation polymorphism analysis (20).
The non-synonymous base changes and the polymorphic variants identified in the CACNA1A gene were named according to Human Genome Variation Society guidelines (http://www.hgvs.org), using RefSeq accession number NM_023035 as a cDNA reference sequence, with nucleotide 283, the A of the ATG initiation codon, corresponding to +1, and NP_075461 as the protein reference sequence. Both sequences correspond to the CACNA1A transcript variant 2. The ATP1A2 reference sequences were NM_000702 for the cDNA and NP_000693 for the protein.
Results
Clinical data
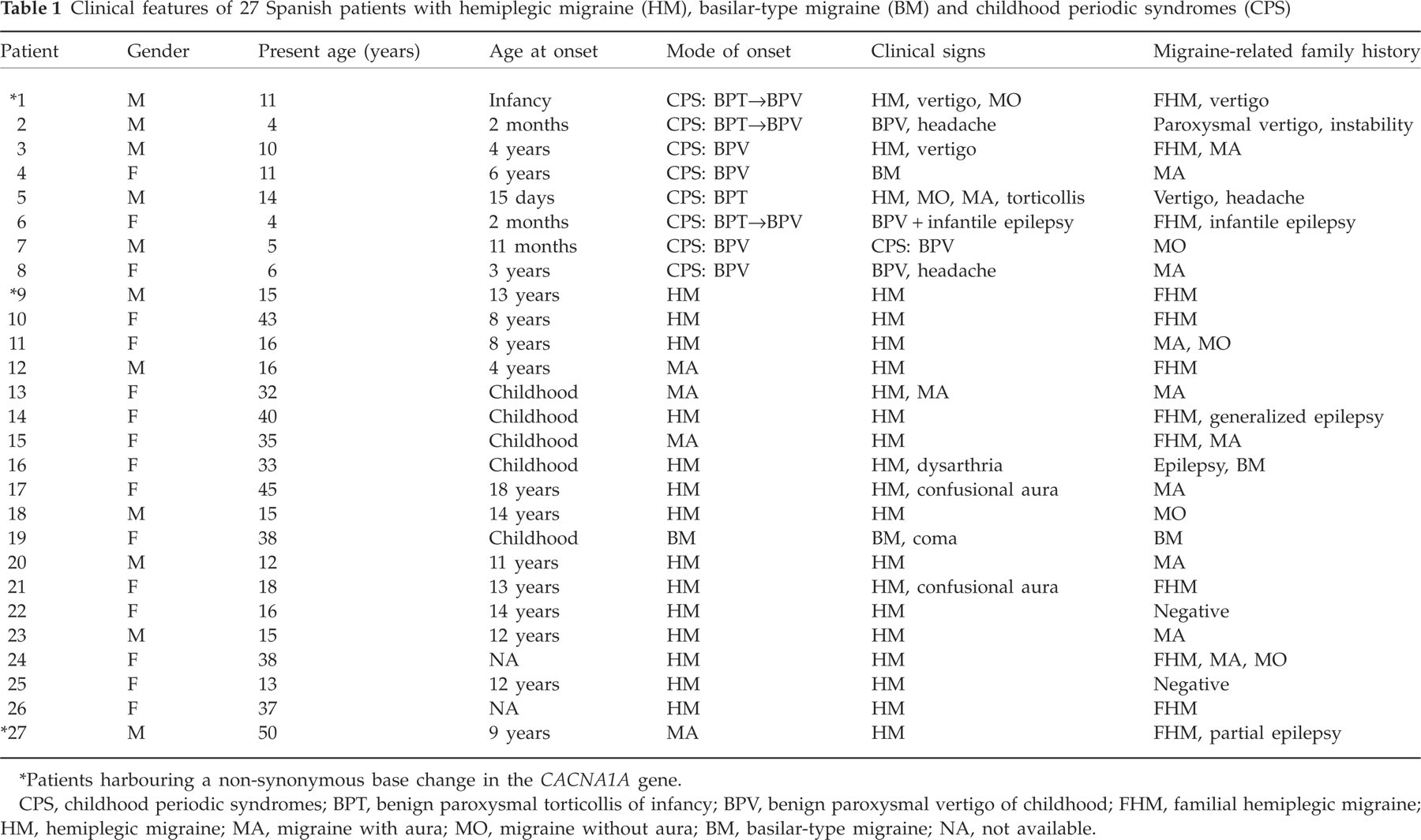
The main clinical features of the 27 patients are shown in Table 1. Episodes of HM were documented in 21 patients, irrespective of the mode of onset. Age at onset ranged from 15 days to 2 months in BPT (n = 4), from 11 months to 6 years in BPV (n = 7) and from 8 to 18 years in HM (n = 21). In the single case presenting as BM, onset was in childhood.
Clinical features of 27 Spanish patients with hemiplegic migraine (HM), basilar-type migraine (BM) and childhood periodic syndromes (CPS)
Patients harbouring a non-synonymous base change in the CACNA1A gene.
CPS, childhood periodic syndromes; BPT, benign paroxysmal torticollis of infancy; BPV, benign paroxysmal vertigo of childhood; FHM, familial hemiplegic migraine; HM, hemiplegic migraine; MA, migraine with aura; MO, migraine without aura; BM, basilar-type migraine; NA, not available.
Eight patients presented as CPS. The main symptom of CPS, torticollis or vertigo, was often associated with pallor, hypotonia, irritability, anxiety, vomiting or crying during the attacks. Of note, three of the four patients that had their onset in early infancy with episodes of BPT subsequently developed BPV, and one of them finally evolved into the HM phenotype. The fourth BPT patient developed HM at age 8 years. Among patients who had their onset in childhood with episodes of BPV (n = 4), one developed HM, one BM and two continued to display BPV at the ages of 5 and 6 years, the latter with accompanying headache. All BPV patients had normal EEG and audiometric testing; clinical screening of vestibular function in school-aged children was also normal.
The remaining 19 patients presented with more typical ‘adult’ phenotypes, including FHM (n = 7), SHM (n = 7), MA (n = 4) and BM (n = 1). All patients with MA went on to develop typical HM attacks. No patient had concomitant ataxia.
Interictally, all 27 patients had normal neurological examinations and all had normal brain magnetic resonance imaging.
Overall, a family history of migraine was present in 23/27 cases, including 18/21 of those developing HM, 2/2 with BM and 3/4 of the younger patients with CPS that had not developed migraine. In 12 families, the affected relatives fulfilled the ICHD-II criteria (17) for FHM.
No other signs suggestive of neuronal channelopathy were noted, except for infantile convulsions that were reported in case 6 and in one of his relatives, and epilepsy in three other unrelated subjects belonging to the families of cases 14, 16 and 27.
Genetic analysis
Two new and one previously cited non-synonymous base changes were identified in three unrelated individuals (cases 1, 9 and 27) after extensive sequencing of the CACNA1A gene in the 27 probands (Table 2). In the remaining 24 patients not bearing CACNA1A variants, sequence analysis of the ATP1A2 gene and exon 23 of the SCN1A gene did not reveal any putative pathogenic change.
Patients and CACNA1A non-synonymous base changes
cDNA and protein sequence numbering are according to Human Genome Variation Society guidelines (http://www.hgvs.org).
cDNA: nucleotide A of the ATG initiation codon corresponds to +1 (nucleotide 283 of the reference sequence NM_023035).
Protein: reference sequence NP_0755461.
In a patient presenting the age-specific sequential phenotypes of BPT, BPV and FHM (case 1) a change from A to G at cDNA nucleotide 3734 in exon 22 was identified, prompting the new gene variant p.Tyr1245Cys, located at domain III, segment 1 (DIII-S1) of Cav2.1. A patient with pure FHM (case 9) was found to harbour the p.Val581Met variant, encoded in exon 13. This was brought about by a G to A substitution at cDNA nucleotide 1741 leading to the substitution of a methionine for a highly conserved valine located at domain II, segment 4 (DII-S4) of the protein. Finally, in another FHM patient (case 27), a change from G to C at cDNA nucleotide 4601 in exon 29 produced the p.Cys1534Ser variant which lies in the intracellular loop between domains DIII and DIV (Fig. 1 and Table 2). The pedigrees of patients 1, 9 and 27 are shown in Fig. 2.

Schematic representation of the human CACNA1A gene (top), the three non-synonymous base changes identified in this study with the corresponding sequence electropherograms (middle) and the encoded protein (bottom). Previously reported familial hemiplegic migraine (FHM) mutations are indicated with numbers on the protein. cDNA and protein sequence numbering are according to Human Genome Variation Society (HGVS) guidelines (http://www.hgvs.org), using NM_023035 as a reference sequence for the cDNA and NP_0755461 for the protein.

Structure of pedigrees of patients with identified gene variants. Affected individuals are denoted by solid symbols. Clinical characteristics are indicated below each individual (HM, migraine with hemiplegic aura; MO, migraine without aura). The CACNA1A variant carrier status is indicated below each index patient.
The presence of these gene variants was confirmed by restriction analysis of the corresponding PCR products, and they were not present in 64 unrelated Spanish non-migraineurs. The residues that were replaced are highly conserved in evolution in paralogous human α1 subunits of Cav2 and Cav1 channels (CACNA1A, B, E, D, F, C and S) as well as in orthologous CACNA1A subunits of cattle (Bos taurus), mouse (Mus musculus), rat (Rattus norvegicus), rabbit (Oryctolagus cuniculus), zebrafish (Danio rerio) and fruit fly (Drosophila melanogaster) (Fig. 3). The conservation is lost only for residue p.Tyr1245 in the human CACNA1D, F, C and S protein (present in Cav1 channels), but not in CACNA1B and E (Cav2 channels), evolutionarily and functionally closer to CACNA1A (21).

Protein alignment performed with clustalW (http://www.ebi.ac.uk/clustalw). The human CACNA1A Val581 and Cys1534 residues are conserved in all the human calcium channel α1 subunits studied (CACNA1B, E, D, F, C and S), whereas Tyr1245 is conserved only in the evolutionarily closer subunits (CACNA1B and E). All three positions are conserved in the orthologous CACNA1A proteins of several organisms. Non-conserved amino acids are indicated in grey. In parentheses, the RefSeq code of each protein. DARE: Zebrafish, Danio rerio; DROME: Fruit fly, Drosophila melanogaster.
No potential pathogenic mutations were found in the coding region of the CACNA1A gene in the remaining 24 probands, some of whom bore polymorphic variants with non-apparent pathogenic effect (Table S3). To our knowledge, four of these changes, c.400-28C>T (intron 2), c.5253-101C>T (intron 34), c.5944-52C>T (intron 40) and c.6840G>A or p.Pro2280Pro (exon 47), are described here for the first time, the latter being located in the coding region of the CACNA1A isoform 2. All these changes were found in several patients and also in a subset of 64 Spanish healthy controls in whom minor allele frequency (MAF) ranged from 0.008 to 0.211. We also found three previously described non-synonymous variants located in exons 19 (rs16022C>G or p.Asp918Glu and rs16023T>A or p.Val993Glu) and 20 (rs16027A>G or p.Ser1105Gly), which were present in several patients and in the general population (Table S3). Three previously described polymorphic variants (rs16041C>T in intron 35, rs16006A>G in exon 6 and c.579G>A in exon 4) were found only in one patient each. Their population frequencies had been studied previously by other authors in several panels of individuals and were found to range between 0.02 and 0.04. One of them, c.579G>A (p.Thr193Thr), was not found in a screening of 64 Spanish non-migraineurs performed by us, although it was present in a set of 50 randomly collected Dutch individuals with a MAF of 0.02 (1).
The 24 individuals not bearing non-synonymous CACNA1A changes did not show ATP1A2 variants either. However, several polymorphic variants were once again identified, including two novel changes in the 5’-UTR region of the gene (c.1-48C>G and c.1-42G>C), both present only in patient 11, and two previously described polymorphic variants (c.749-43G>C in intron 7 and rs2070701G>A in the 3’-UTR region), also found in only one patient each (Table S3). The presence of the p.Gln1489Lys SCNA1A missense mutation was ruled out in these 24 individuals.
Discussion
We have reported two novel and one previously cited non-synonymous base changes in the CACNA1A gene in a set of 27 unrelated, predominantly familial, Spanish cases with different migraine variants (HM, BM, CPS). To our knowledge, this is the first molecular analysis of the CACNA1A, ATP1A2 and SCN1A genes to have been conducted in Spanish patients.
Molecular genetics of HM
In our series, the ratio of three cases bearing CACNA1A changes out of 11 pure FHM cases (27.3%) is roughly in line with figures reported elsewhere (e.g. 4/12 (22) or 6/42 (3)), but contrasts to the widely accepted notion that mutations in the CACNA1A gene account for more than half of FHM cases, reviewed in (2). However, a more detailed review of the literature reveals that these high percentages were recorded only when mutational screenings included FHM families selected on the basis of their putative linkage to the CACNA1A locus on 19p13 or on that of the occurrence of concomitant cerebellar ataxia. Thus, 15 mutations were found in 16 FHM families with cerebellar signs and positive linkage to CACNA1A (22) and five out of six FHM families with previous positive linkage, two of them with cerebellar signs (1). The proportion of patients with CACNA1A mutations is much lower in the case of SHM, and always < 5% in the absence of cerebellar signs (1, 23–25). In our study we found no CACNA1A mutation among the 10 SHM cases without cerebellar signs, despite the fact that seven of them displayed a familial history of (non-hemiplegic) migraine.
It is of note that no ATP1A2 non-synonymous base changes were found in our series. This is in contrast with previous studies that reported percentages of 42% (11/26) (26), 17% (1/6) (27) and 7% (3/42) (3) for ATP1A2 mutations in FHM patients without mutations in the CACNA1A gene. However, no ATP1A2 mutations were found in a study including 19 FHM and seven BM patients (25). Mutations in SCN1A, the only other gene linked to FHM and involved in several forms of epilepsy, would appear to be a rather unusual cause of FHM. Indeed, > 160 mutations have been identified in SCN1A (Human Gene Mutation Database, http://www.hgmd.cf.ac.uk) (28) compared with just one in FHM (13). Finally, at least one other FHM locus has been mapped to 1q31, but the underlying gene awaits identification (29).
The fact that only three CACNA1A non-synonymous base changes have been identified in 27 patients with HM, BM or CPS after an exhaustive screening of CACNA1A and ATP1A2 may be explained by the high level of genetic heterogeneity in this group of paroxysmal conditions. Nevertheless, it is also possible that a low number of mutations have remained unidentified in the genes studied, including changes in introns or in the regulatory regions, or gross alterations that may be undetectable by PCR.
Childhood periodic syndromes and CACNA1A
In this report we have described eight patients that presented with a CPS phenotype in infancy or childhood. The various CPS phenotypes have generally been considered paediatric equivalents of migraine, and three of them, BPV, CV and abdominal migraine, are recognized by the IHS as migraine equivalents in infancy or early childhood. A fourth phenotype, BPT, awaits validation as CPS or migraine precursor. BPT might very well be the earliest manifestation of migraine in life. In fact, our patient 1 presented with torticollis episodes during the neonatal period, similar to two cases reported elsewhere (30, 31). Our patient was found to bear the p.Tyr1245Cys variant in the CACNA1A gene, and his clinical course reflects the changing, age-specific phenotypes associated with CACNA1A dysfunction, i.e. BPT, BPV and FHM. This sequence in the leading clinical manifestations, which may at times overlap, was seen in three of our four BPT patients and has also been suggested by other authors (32–35). Our results suggest a common genetic background for some forms of CPS and FHM. A previous study (16) has reported a patient with BPT featuring interictal ataxia and who belonged to one kindred with FHM and ataxia linked to a CACNA1A mutation. However, the authors did not disclose the genotype of the probandus. BPV, in turn, is the most common cause of childhood vertigo without ear disease or hearing loss (36). Classic BPV begins between 1 and 4 years of age, and further evolution towards MA is observed in > 50% of cases (37, 38). Among our seven BPV cases, three have developed classical migraine phenotypes by age 10: FHM in two cases and BM in one. The remaining four patients are < 7 years old; two of them complain of recurrent headaches.
Description of identified CACNA1A non-synonymous base changes
Several findings point to a causative role of the three variants identified in the CACNA1A gene, p.Val581Met, p.Tyr1245Cys and p.Cys1534Ser, although the eventual demonstration of their pathogenicity in FHM will require functional studies.
The p.Val581Met variant (patient 9) involves a residue located within the transmembrane S4 segment of the DII domain (DII-S4), and the change is predicted to shorten a α helix, as determined by the PSIPRED software (http://www.psipred.net) (39). This segment probably represents the voltage sensor of the channel, in which most mutations have been described. All the mutations reported to date in S4 segments, except for that reported here, are substitutions of a conserved arginine (192, 195, 583, 1347, 1661, 1664 and 1667), and the majority of them have been associated with the combination of hemiplegic migraine and episodic ataxia or cerebellar signs.
The p.Tyr1245Cys variant (patient 1) affects the transmembrane DIII-S1 segment. No other missense/nonsense changes have previously been described in the S1 segment of any domain of the α1A subunit of the CaV2.1 channel in any patient with FHM. This may argue against its involvement in the disease, although the total number of FHM mutations that have been described in the gene so far (around 20) is still small.
The third gene variant identified, p.Cys1534Ser (patient 27), has been described in a previous report in three relatives with FHM, two of them with concomitant episodic ataxia (40). However, the presence of this variant in the general population was not assessed, and its putative structural/functional relevance not discussed. Prediction of the protein secondary structure indicates that the amino acid change produces a shortening of a α helix, which might have functional consequences for the channel. The mutation involves a residue located in the intracellular loop between domains DIII and DIV, where, to our knowledge, no other mutations have been described in FHM. Interestingly, the CACNA1A protein is structurally very similar to SCN1A, and the only FHM mutation (p.Gln1489Lys) described so far in the latter is, like Cys1534Ser, also located in the DIII-DIV linker.
Other evidence supports the relation of these three non-synonymous base changes with the disease phenotype. First, the residues involved are highly conserved in evolution, both at the intraspecific (human CACNA1A, B, C and E subunits) and interspecific levels (CACNA1A subunit of human, cattle, mouse, rat, rabbit, zebrafish and fruit fly), indicating functional/structural relevance (Fig. 3). Second, we did not detect the presence of these gene variants in a screening of 64 healthy Spanish individuals in whom migraine had been specifically excluded, and the changes are not present in the public single nucleotide polymorphism databases, indicating that they are not polymorphic variants. Third, no other molecular alterations were identified within the gene after the analysis of the whole coding region and the exon–intron boundaries, including splice sites and branch points. Unfortunately, family members were unavailable for cosegregation analysis between the disease phenotype and the identified variants, which may had provided additional clues about their putative causative role.
Conclusions
Mutational analysis of ion channel genes is often expensive and time-consuming, but it is conceivable that some clinical clues might help guiding molecular diagnosis in HM. Thus, episodic or permanent cerebellar ataxia or trauma-induced episodes should prompt investigation of CACNA1A mutations (FHM1). Alternating hemiplegia or BM may suggest a defect in ATP1A2 (FHM2). Concomitant epilepsy suggests mutations in SCN1A (FHM3), although absence of epilepsy has been described in FHM1 and benign familial infantile convulsions in FHM2.
On the basis of the results reported here and previous molecular genetic studies, we hypothesize that, although the list of FHM-causative genes is expected to grow, a substantial number of cases may follow more complex patterns of inheritance, similar in this respect to other migraine variants.
Acknowledgements
The authors wish to thank all the patients and family members for their cooperation and J. Artigas, M. Pineda, A. Rodríguez and M. Galván for patient referral. J. M. Fernández is acknowledged for helpful suggestions in the preparation of the manuscript, and M. Ribasés for technical assistance. This study was supported by the Spanish Ministry of Education and Science (SAF-2000/197 and SAF-2003/04704), Red Española de Ataxias, Fondo de Investigación Sanitaria, Spain (G03/056 and PI052129), Instituto de Salud Carlos III, Spain (PI061073, PI050996), Fundació la Marató de TV3 (061330) and AGAUR (2005SGR00848). E.C-L. was a recipient of a FPI scholarship of the Ministerio de Ciencia y Tecnología, Spain. R.C. was funded by the Institut de Recerca Vall d'Hebron.