
Abstract
The aim of this study was to delineate any dysfunction of neuromuscular transmission (NMT) by single-fibre electromyography (SFEMG) in some rare types of migraine. Recent studies have shown subclinical dysfunction of NMT in migraine with aura and cluster headache by using SFEMG, whereas another recent study has shown NMT to be normal in familial hemiplegic migraine (FHM) with CACNA1A mutations. Thirty patients with rare primary headache syndromes [18 with sporadic hemiplegic migraine (SHM), six with FHM and six with basilar-type migraine (BM)] and 15 healthy control subjects without any headache complaints underwent nerve conduction studies, EMG and SFEMG during voluntary contraction of the extensor digitorum communis muscle. Ten to 20 different potential pairs were recorded and individual jitter values calculated. The results obtained from patient groups were compared with those from the normal subjects. Of 600 individual jitter values of the patients, 27 (4.5%) were abnormally high, whereas only 3/205 (1.5%) jitter values from normal subjects were abnormal. Abnormal NMT was found in 4/30 (13.3%) patients (three SHM and one BM), but in none of the control subjects. Only in SHM patients was the number of individual abnormal jitter values slightly but significantly different from normal controls. The present study demonstrates that subclinical NMT abnormality is slightly present in only SHM and BM patients, but not in FHM patients.
Keywords
Introduction
Abnormal neuromuscular transmission (NMT) without a clinical correlate has been detected by stimulation single-fibre electromyography (SFEMG) in migraine with aura (MA) (especially in individuals with prolonged aura) and migraine without aura (MoA) (1–4). We have recently shown pronounced abnormalities in NMT by SFEMG in a small group with episodic cluster headache (5).
The first SFEMG studies on migraine suggested that P/Q Ca2+ channel abnormality due to CACNA1A gene mutation had a role in the pathophysiology of NMT abnormality and MA, especially with non-visual aura (1–3). However, a SFEMG study on familial hemiplegic migraine (FHM) patients, half of whom had CACNA1A mutations and were expected to have more extensive NMT abnormality due to proven genetic abnormality, has disclosed that tests for NMT were not significantly different from healthy control subjects and the authors concluded that NMT was normal in FHM (6). A subsequent study on migraine patients, including hemiplegic and basilar type migraine, provided similar results to previous ones (4). However, except for the study on FMH patients (6), none of these studies gave definite ratios for the incidence of NMT abnormality in migraine subgroups such as sporadic hemiplegic migraine (SHM), FHM and basilar-type migraine (BM).
In this study, we aimed to search for any NMT abnormality in patients with some rare types of migraine.
Methods
Subjects
Thirty patients (21 women, nine men; age 17–54 years, mean 35.2 ± 11.7 years) with FHM or BM and 15 age-matched healthy control subjects (nine women, six men; aged 16–55 years, mean 35.5 ± 10.7 years) were included, after giving written consent. There were 18 patients with SHM, six with FHM and six with BM. The subjects were diagnosed according to the criteria of International Headache Society (7) between 2000 and 2004 in headache out-patient clinics of three referral centres. The patients who had applied before 2004 were reconsidered for the SHM diagnosis which took its place for the first time in the International Classification of Headache Disorders, Second Edition (ICHD-II) in 2004. The local ethics committee of Istanbul University approved the study.
All patients and healthy controls underwent a detailed neurological evaluation. None of the normal subjects had a primary headache disorder and none of study subjects had any neurological complaints or findings suggestive of any neuromuscular disorder. No electrophysiological examinations, including SFEMG, were performed during a migraine attack in any of the patients. None of the patients was under prophylactic treatment during the SFEMG investigation or used any drugs which could have a potential effect on the NMT. Six out of 30 (three SHM, three BM; 20%) patients used tobacco and three (one SHM, two BM; 10%) used alcohol within the limits of social drinking.
All study subjects had normal neurological examination out of the attack period. All patients and control subjects had normal nerve conduction studies and concentric needle EMG studies of the extensor digitorum communis (EDC) muscle.
Electrophysiological study
Motor nerve conduction studies of the median, ulnar, peroneal and tibial nerves, sensory nerve conduction studies of the median, ulnar and sural nerves, and concentric needle EMG and SFEMG of the EDC muscle were performed. During all the electrophysiological examinations, the skin temperature was maintained above 32°C in the upper extremity and above 31°C in the lower extremity.
Single-fibre EMG was performed during voluntary contraction of the EDC muscle, and a single-fibre electrode (22596; Medelec, Old Woking, UK) was used for recording. A Keypoint electromyograph (version 3.2; Denmark) was used for recording, saving data, and analysis. The low-cut filter was 500 Hz and high-cut filter was 10 kHz for the SFEMG recording. Only potentials with a stable shape, a rise time of <0.3 ms and amplitude of >200 µV were accepted for jitter analysis. For each jitter analysis, 50–100 consecutive traces were recorded (8). For each patient, 20 different single-fibre potential pairs were recorded and jitters of these potential pairs were calculated. In control subjects, 10 (nine subjects), 15 (one subject) or 20 (five subjects) different single-fibre potential pairs were recorded. The mean consecutive difference (MCD) and mean sorted difference (MSD) were calculated. The calculated MCD was accepted as ‘jitter’. If the MCD/MSD ratio was >1.25, MSD was used instead of MCD as the jitter value. The number of individual abnormal jitter values exceeding the upper limit of normal was calculated for each subject. The upper normal limit of the individual jitter was 55 µs. The number of abnormal individual jitter values was taken into account for the interpretation of the SFEMG data. The range of normal values used for individual jitter results were derived from a multicentre study (9). If a subject had >10% individual abnormally high jitter values out of all individual jitter values (e.g. >2 out of 20), NMT was labelled as ‘abnormal’.
Statistical analysis
Mean values of jitter and abnormally high individual jitter values were calculated for the two groups (patients and normal groups). To compare the individual jitter values of the groups, parametric comparison tests (unpaired t-test and one-way
Results
Table 1 summarizes the demographic characteristics and the NMT results of the patients.
The demographic characteristics of patients and controls
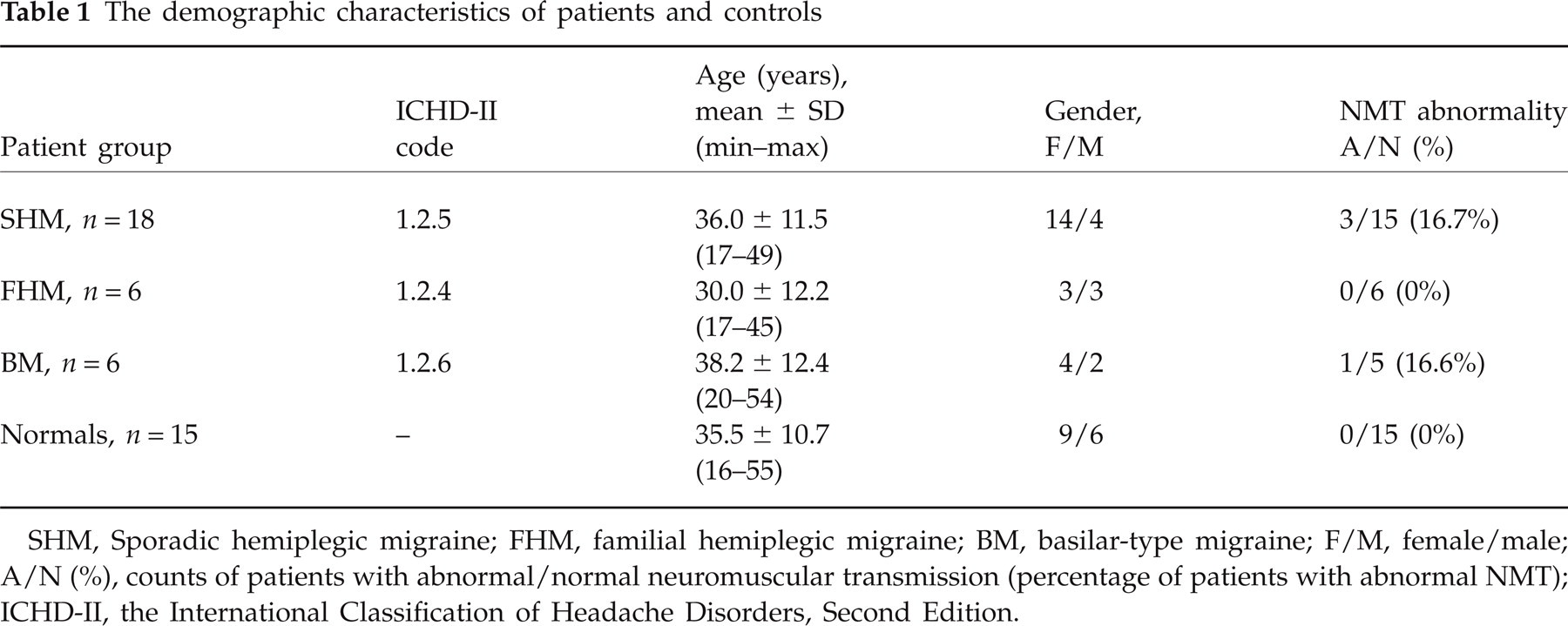
SHM, Sporadic hemiplegic migraine; FHM, familial hemiplegic migraine; BM, basilar-type migraine; F/M, female/male; A/N (%), counts of patients with abnormal/normal neuromuscular transmission (percentage of patients with abnormal NMT); ICHD-II, the International Classification of Headache Disorders, Second Edition.
A total of 360 individual jitter values were obtained from 18 SHM patients, 120 from six FHM patients, 120 from six BM patients and 205 from 15 age-matched normal control subjects. The mean value of individual jitters was 26.9 ± 10.7 µs (range 9–98 µs) in normal control subjects, whereas the mean was 27.0 ± 15.2 µs (range 7–140 µs) for SHM patients, 26.9 ± 11.2 µs (range 9–62 µs) for FHM patients and 29.9 ± 24.4 µs (range 8–162 µs) for BM patients (Fig. 1). Neither parametric nor non-parametric comparison tests showed any statistically significant difference between the individual jitter values of the groups.
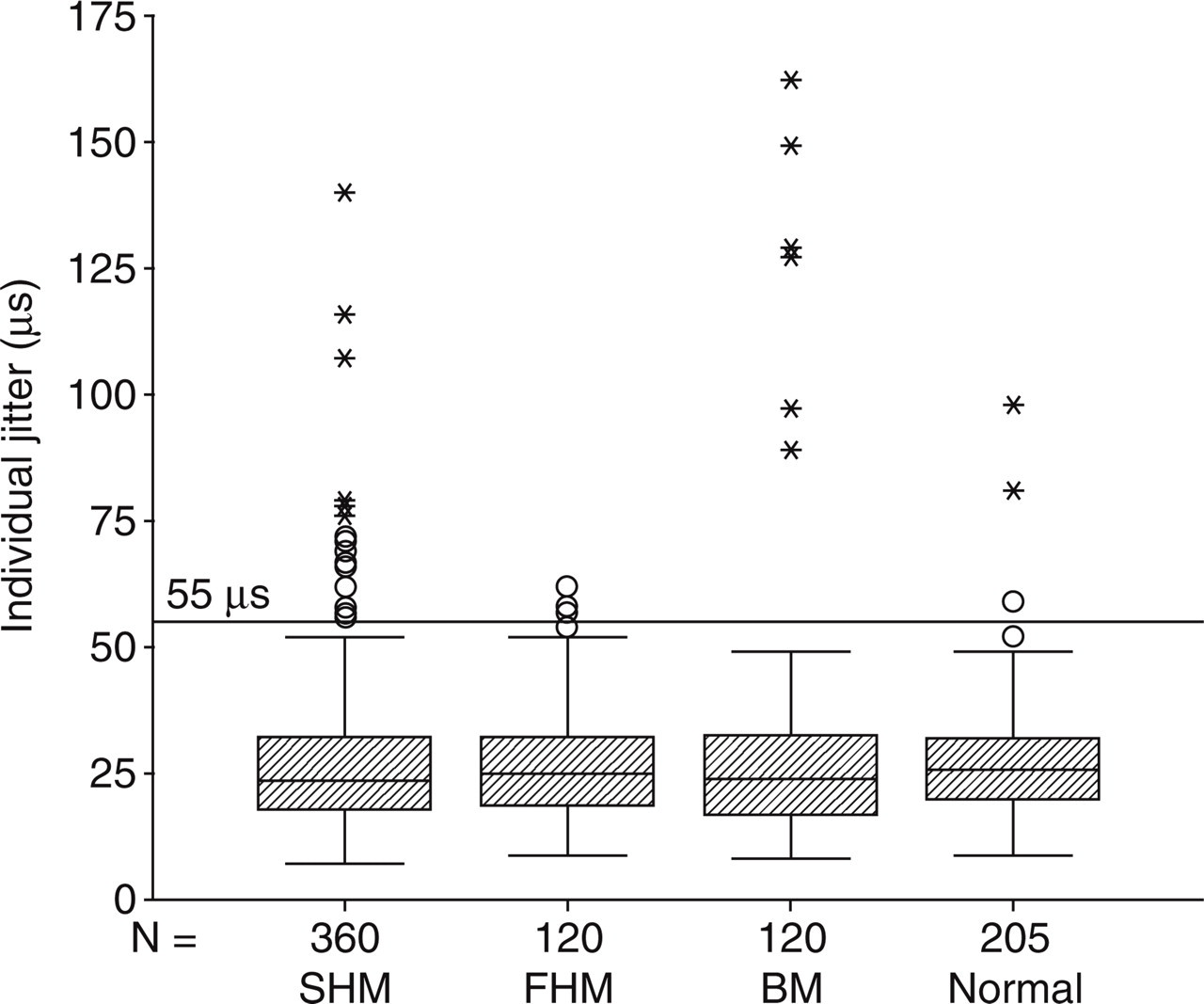
Distribution of individual jitter values in patients and normal controls. SHM, Sporadic hemiplegic migraine; FHM, familial hemiplegic migraine; BM, basilar-type migraine.
Of 205 individual jitter values of the normal controls, only three (1.5%) were abnormally high, whereas 17 (4.7%) of 360 jitters from SHM patients, four (3.3%) of 120 from FHM patients (not significantly different from controls) and six (5%) of 120 jitters from BM patients were abnormally high. The count of abnormal individual jitter values of SHM patients was significantly higher than that of controls (χ2 Fisher's one-sided exact test P = 0.032), whereas FHM and BM groups were not significantly different from controls.
SFEMG investigation revealed abnormal NMT in three (16.7%) SHM patients and one (16.7%) BM patient, whereas none of the control subjects and FHM patients had abnormal NMT (Table 1). In none of the patient groups was the count of the patients with abnormal NMT significantly different from that of controls by χ2 test.
There were no apparent differences in headache symptoms (intensity and duration of attack) or clinical features (accompanying aura symptoms) between the patients with abnormal or normal NMT.
Discussion
Recent studies have shown that a minority of migraineurs have mild subclinical NMT abnormalities, for unknown reasons (2–4). Ambrosini has reported that a total of 17 of 62 migraine patients had pronounced SFEMG abnormalities (3). All except one had MA, six had prolonged aura and the majority had, in addition to the visual scotoma, other aura symptoms such as sensory/motor disturbances, dysphasia and loss of balance (3). None of them had the diagnosis of FHM or BM and six had motor aura symptom. The number of patients with motor aura symptom and also NMT abnormality was not given in the report of Ambrosini (3). Domitrz has reported that half (6/12) of the patients with hemiplegic migraine, paraesthesias and speech disturbances during aura, or persistent aura without infarction (of these 12 patients, four had SHM, four had FHM, one had BM) had NMT abnormality, without giving the NMT abnormality ratio within these subgroups such as SHM, FHM and BM (4). These studies have suggested that genetically determined P/Q Ca2+ channel abnormality may play a role in underlying mechanism of both NMT abnormality and MA. However, in a SFEMG study of 12 FHM patients (six with CACNA1A mutation, FHM1), Terwindt has found NMT abnormality in only two patients (all with CACNA1A mutation), whereas one healthy control subject also had the same abnormality, and has suggested that this mutation has no role in pathology (6). Our study has shown that NMT was also abnormal in some patients with SHM and BM, which are rare types of MA. This result could be interpreted as a functional abnormality of neuromuscular junction constituents in a group of migraine syndromes whose pathophysiology relies on both genetic and environmental factors (10).
FHM, SHM and BM are phenotypically similar subtypes of MA, differentiated only by motor symptoms, which are absent in BM, and by the presence of a positive family history of FHM (11). A novel mutation in the ATP1A2 gene (R548H), which is also responsible for FHM, has been detected in members of a family with BM, suggesting that BM and FHM may be allelic disorders (11). There is ongoing controversy about the place of BM, but there is no firm clinical, epidemiological or genetic evidence that BM is an independent disease entity different from MA (12, 13). FHM type 1 patients have mutations of the CACNA1A gene encoding for the pore-forming subunit of the P/Q Ca2+ channels (1). These ionic channels are responsible for acetylcholine release at the neuromuscular junction, thus their impaired functioning could lead to NMT abnormalities (1). In fact, these junction abnormalities have been demonstrated in mutant mice (14, 15) and in patients suffering from episodic ataxia type 2 (EA-2) (16, 17), which is an allelic disease of FHM1 (16). Furthermore, normalization of the abnormal values in SFEMG was obtained in two migraine patients treated with acetazolamide, currently used to treat FHM1 and EA-2 patients (18). Similar results were obtained in a subsequent study by another group, who found mild SFEMG abnormalities in two of five patients with non-hemiplegic migraine (19). However, the current study and another study of 12 FHM patients with CACNA1A mutations (6) do not support this hypothesis of P/Q Ca2+ channel abnormality due to CACNA1A mutations. Although severe aura symptoms (such as sensori-motor symptoms, language impairment in five FHM patients) or prolonged duration of the aura (such as the presence of hemiplegia for 4 days in one FHM patient) were seen in our patients, SFEMG was normal in all FHM of our patients.
Compared with general population, SHM probands had no increased risk of MoA, but a greatly increased risk of typical MA (20, 21). This epidemiological finding, as well as our electrophysiological data showing NMT abnormality in SHM similar to other patients with MA, may highlight genetic overlaps between the migraine subtypes, and raises some hopes that the SFEMG abnormality might serve as a marker for a specific, currently unknown genetic defect.
In conclusion, we have shown abnormal NMT in three of 18 patients with SHM, in one of six patients with BM and in none of six patients with FHM by using SFEMG. No prominent clinical differences were found between patients with abnormal and normal NMT.