
Abstract
The aim of this study was to investigate the involvement of the CACNA1A and ATP1A2 gene in a population-based sample of sporadic hemiplegic migraine (SHM). Patients with SHM (n = 105) were identified in a nationwide search in the Danish population. We sequenced all exons and promoter regions of the CACNA1A and ATP1A2 genes in 100 patients with SHM to search for possible SHM mutations. Novel DNA variants were discovered in eight SHM patients, four in exons of the CACNA1A gene and four in exons of the ATP1A2 gene. Six of the variants were considered non-pathogenic. The causal role of the two remaining DNA variants is unknown until functional studies have been made or independent genetic evidence is discovered. Only very few DNA variants were identified in 100 SHM patients, and regardless of whether the identified variants are causal the CACNA1A and ATP1A2 genes are not major genes in SHM.
Introduction
Familial hemiplegic migraine (FHM) is an autosomal dominant subtype of migraine with aura (MA), where the aura includes some degree of motor weakness and where at least one first- or second-degree relative has identical attacks. Mutations have been reported in the CACNA1A (FHM1) (1–3), the ATP1A2 (FHM2) (3–10) and the SCN1A (FHM3) gene (11), and mutations in one of these genes is found in 14% of population-based and 70% of selected families with FHM.
Sporadic hemiplegic migraine (SHM) is hemiplegic migraine in families with one affected member. Only case reports and small selected series of patients with SHM have been examined for mutations in the CACNA1A and ATP1A2 genes.
In order to describe the genetic spectrum of SHM, we sequenced all exons and promoter regions of the CACNA1A and ATP1A2 genes in 100 patients with SHM identified from a nationwide search in the general Danish population.
Methods and materials
Search for patients with SHM
A systematic search for patients with SHM was performed employing three different search strategies in the entire Danish population of 5.2 million. The discovery of patients, search strategy, telephone interview, participation/non-participation rates and further details about material have been reported previously (12), and the clinical characteristics of the SHM patients have been reported elsewhere (13).
Thus, a total of 105 patients with SHM from 105 different families were available for the present study. These families were selected for a sequence analysis of the CACNA1A and ATP1A2 genes. After obtaining informed consent according to the Declaration of Helsinki, venous blood samples were collected from SHM-affected patients and their available unaffected parents. A total of 100 of the 105 SHM patients participated by giving blood samples for genetic testing. Five SHM patients refused to participate for unspecified reasons. The project was approved by the Danish Ethics Committees.
Patient material
One hundred SHM patients were screened for mutations in both the CACNA1A and ATP1A2 genes. In affected patients with a DNA variant, additional available family members (parents and other unaffected relatives) were screened for the identified DNA variant. Conservation of amino acid residue in 17 vertebrate species was investigated (14). An amino acid residue is regarded as conserved if it is the same in all 17 species. These species are human, chimp, macaque, mouse, rat, rabbit, dog, cow, armadillo, elephant, tenrec, opossum, chicken, frog, zebrafish, tetraodon and fugu. The number species with the amino acid conserved are indicated relative to the number of species evaluated (Table 1). Amino acids are numbered according to location in protein sequence of CAC1A_Human (Swiss-Prot accession O00555) and AT1A2_Human (Swiss-Prot accession P50993).
DNA variants identified in the ATP1A2 and CACNA1A genes

Amino acids are numbered according to location in protein sequence of CAC1A_Human (Swiss-Prot accession O00555) and AT1A2_Human (Swiss-Prot accession P50993).
Conservation of amino acid residue in 17 vertebrate species (14). These species are human, chimp, macaque, mouse, rat, rabbit, dog, cow, armadillo, elephant, tenrec, opossum, chicken, frog, zebrafish, Tetraodon and Fugu. The number of species with the amino acid conserved is indicated relative to the number of species evaluated. For E2080K 13 species have glutamic acid (E) and two species have a change to the similar aspartic acid, indicating a tendency for conservation, as indicated by the number 15.
All the amino acid changes described here are in protein loop regions with extracellular (Ex) or intracellular (In) location or in the C- or N-terminus.
The number of unaffected relatives of sporadic hemiplegic migraine proband with the codon change out of the number of relatives screened. One parent has the codon change for all cases tested.
Unaffected controls with the codon change. In a separate study of Danish familial hemiplegic migraine (FHM) families (3), one unaffected member of family 6034 (with known R583Q mutation) was identified with the Y9N amino acid change (not reported).
A population control group consisting of 92 unrelated persons without migraine, diagnosed in an extensive interview by a physician, was included for the sequence analysis of exons that revealed DNA variants in patients.
Sequence analysis
The 48 exons of the CACNA1A (19p13) and the 23 exons of the ATP1A2 (1q23) gene were sequenced, including promoter and flanking intron sequences, using primers as previously described (15). Polymerase chain reaction (PCR) and cycle sequencing primers were designed by WinSeq1.6, a deCODE software based on the Primer3 software (16). PCR amplifications were set up on Sciclone ALH 300 and run on MJR TetradTM. PCR products were verified for correct length on agarose gel before being purified using AMPureTM from Agencourt (Beverly, MA, USA). Cycle sequencing reactions were set up on Sciclone ALH 300, run on MJR TetradTM, and excess dye terminators were removed using CleanSEQTM from Agencourt. Amplimers were sequenced directly on an Applied Biosystems 3730 Capillary DNA Sequencer using an ABI PRISM® Fluorescent Dye Terminator System (Perkin-Elmer, Foster City, CA, USA). The sequence analysis was conducted with Clinical GenomeMinerTM 1.5, a deCODE software based on assembly comparable to Consed (17).
Results
In the CACNA1A gene four previously unreported DNA variants in coding exons were found, causing a change in amino acid (Table 1).
The E2080K amino acid substitution affects a somewhat conserved glutamic acid located in a C-terminus region of the protein. Thirteen species had glutamic acid (E) and two species had a change to the similar aspartic acid, indicating a tendency for conservation in 15 out of 17 species (Table 1). However, two species (armadillo and tenrec) lacked this part of the protein altogether. The H2481Q amino acid variant was not found in 92 controls, but the healthy mother was a mutation carrier. This amino acid changed a non-conserved amino acid in the C-terminus region of the protein and it was found in three unrelated population controls. The P897R and P2479L substitutions affect amino acids that are not conserved among species, but neither were found in 92 controls. Both were found in an unaffected parent. The P897R substitution changes a non-polar proline to a basic polar arginine in the intracellular loop of the protein, and the P2479L substitution changes a non-polar proline to a non-polar leucine in the C-terminus region of the protein. None of the four amino acid changes are located close to the location of known FHM mutations (Fig. 1).
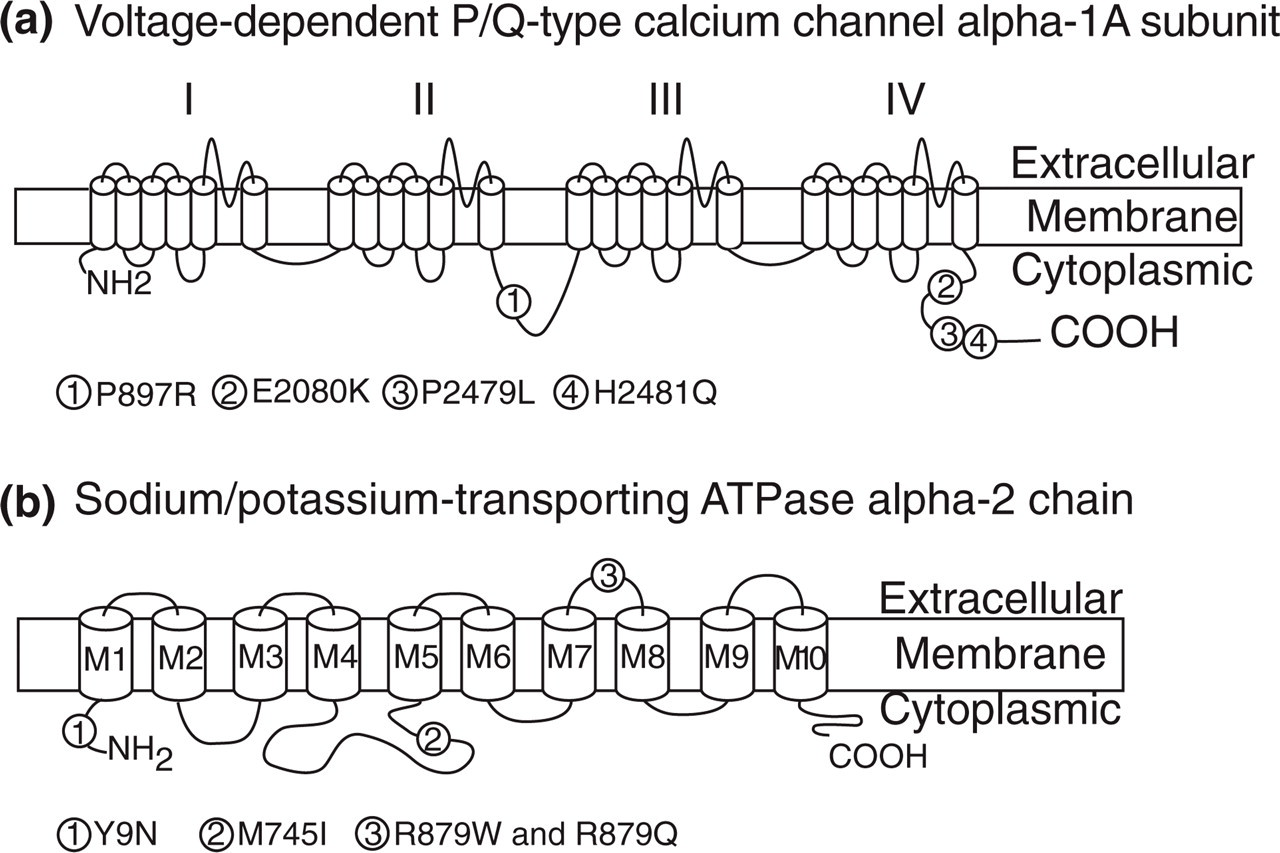
The location of identified DNA variants in the CACNA1A and ATP1A2 genes.
In the ATP1A2 gene we found four additional DNA variants in coding exons, causing a change in amino acid (Table 1). The Y9N amino acid substitution affects a conserved tyrosine in a N-terminus region of the protein. It changes an aromatic tyrosine into a smaller asparagine. This variation was not detected in any of the 92 controls tested, but we have previously observed it in an unaffected member of FHM family 6034 with the R583Q mutation (3). The healthy mother was a carrier of this DNA variation. The M745I amino acid variation is located in an intracellular loop of the protein and changes a conserved non-polar methionine to a non-polar isoleucine. This amino acid change is located close to previously reported FHM mutations (Table 1). The proband's mother carried the M745I variant, but neither of the maternal parents. The R879Q and R879W amino acid substitutions affect the same non-conserved arginine in an extracellular loop of the protein. Neither variation was detected in the 92 controls, but both were found in more than one unaffected family member (including an unaffected parent).
The clinical characteristics of SHM patients with the described DNA variation in either CACNA1A or ATP1A2 are shown in Table 2. The affected and unaffected family members and their DNA variation carrier state are shown in Table 3.
Clinical characteristics of sporadic hemiplegic migraine (SHM) patients with CACNA1A and ATP1A2 DNA variants

AO, Age at onset of SHM; +, symptom present in all or most attacks. −, symptom never present; MA, migraine with aura; MoA, migraine without aura.
The sporadic hemiplegic migraine (SHM) family members in the mutation screen and identified DNA variants

The proband of each family is underlined. Other family members have the same inital four digits as the proband.
F, female; M, male.
Discussion
The present study is the first population-based study of SHM and the largest sample of SHM patients screened for mutations in genes that cause FHM. We identified eight previously unreported DNA variants in the CACNA1A and ATP1A2 genes, four in each (Table 1). All identified variants except the ATP1A2 M745I variation are in protein regions never previously implicated in disease. Based on conservation among species, properties of the amino acids and screening of healthy family members and population controls, we consider six of the DNA variants as most likely to be non-pathogenic variants, since they affect non-conserved amino acid residues or are found in controls or in many unaffected relatives.
Since functional studies, which are crucial to assess whether a DNA variant is causal (especially in case of sporadic patients), are lacking for the described amino acid variants, it is not possible to claim a role for these changes in causing SHM, including the last two DNA variants (E2080K DNA variant in the CACNA1A gene and the M745I DNA variant in the ATP1A2 gene). Furthermore, it cannot be ruled out that SHM patients could have mutations in non-coding regulatory regions of the CACNA1A and ATP1A2 genes, not including the screened promoter regions.
In the present study, a total of 92/100 SHM patients did not have a change in DNA sequence in the CACNA1A or ATP1A2 genes. Thus, most SHM patients do not have mutations in these genes, which is in line with previous literature (2, 8, 18–21). One of these studies, comprising 27 patients with SHM, has shown that only two patients had CACNA1A mutations (18): a T666M mutation in a patient with SHM and cerebellar ataxia and a R583Q mutation in a patient with SHM but without cerebellar ataxia. In another study, three patients with SHM and cerebellar symptoms were analysed for mutations in the CACNA1A gene. In these patients two mutations were found, a T666M mutation in a patient with SHM and cerebellar ataxia and a Y1384C mutation in a woman with mental retardation, SHM, coma, seizures and permanent cerebellar ataxia and atrophy (19). In other studies, six patients with SHM without cerebellar symptoms were screened for mutations in the CACNA1A gene, but no mutations were found (2, 21). In the ATP1A2 gene, so far only one mutation has been found in a patient with SHM (R383H). In that study only one of 24 patients with SHM had a mutation (8). Thus, worldwide three different missense mutations in the CACNA1A gene and one different missense mutation in the ATP1A2 gene have now been demonstrated to cause SHM, and only two SHM mutations (T666M and R583Q) have also been found in FHM. Thus, the present study supports the previous data, that the CACNA1A and ATP1A2 genes are not major genes in SHM.
This suggests that SHM could be an aetiologically heterogeneous disorder. Among SHM patients, several groups of patients probably exist. First, patients in whom the disease is due to a mutation in one of the FHM genes. In such patients the disease appears as non-familial, either because the patient has a de novo mutation in one of the FHM genes, or because of incomplete penetrance in other family members carrying the mutation, or finally because there is a false paternity and the real biological father has FHM. Second, in most SHM patients with mutations in the CACNA1A and ATP1A2 genes, the mutations are different from those found in FHM. An explanation may be that the penetrance of these mutations is lower than the FHM mutations in the same genes. However, the great majority of SHM patients have no mutation in two (CACNA1A and ATP1A2) of the known FHM genes. This poses the question, is SHM a genetic disorder? Previously we have shown that SHM occurs with approximately the same prevalence as FHM (12). SHM has an age at onset of 20 years (95% confidence interval 18.3–22.2), which is not significantly different from FHM (22). When comparing the aura and headache characteristics of SHM with those of FHM (22), similar clinical characteristics regarding frequency of each aura symptom, combinations of aura symptoms, sequence of the various aura symptoms, the gradual progression in time and total duration of each aura symptom, headache characteristics and accompanying symptoms have been found, and the aura was almost always followed by headache in both conditions (13, 22). Furthermore, when studying the relation between SHM and basilar migraine, 72% of patients with SHM fulfilled the criteria for basilar migraine during SHM attacks, the same as in FHM attacks (22, 23). The many clinical similarities between FHM and SHM support the hypothesis that SHM is very likely to be a genetic disorder. SHM could be caused by mutations in different, still unknown specific SHM genes with a low penetrance compared with mutations in the FHM genes. However, the great majority of SHM patients do not have mutations in any of the FHM genes, and we therefore suggest that some SHM patients could share common mechanisms with other varieties of migraine, such as migraine with typical aura, and just happened to have a couple of severe attacks with motor weakness. Thus, SHM both in its pure form and co-occurring with typical MA, could be caused by genetic factors that might also be involved in typical MA.
Footnotes
Competing interests
None to declare.
Acknowledgements
We thank the patients with sporadic hemiplegic migraine and their unaffected relatives who agreed to participate. We also thank our colleagues for their excellent collaboration. This study was supported by grants from the Cool sorption Foundation of 1988, the Foundation for Research in Neurology, the Danish Headache Society, the A.P.M⊘ller Foundation for Advancement of Medical Science, the Novo Nordisk Foundation, the IMK-Almene Foundation, Ms Else Torp Foundation and deCODE Genetics.