
Abstract
Ethanol stimulating transient receptor potential vanilloid 1 (TRPV1) on primary sensory neurons promotes neurogenic inflammation, including calcitonin gene-related peptide (CGRP)-mediated coronary dilation. Alcoholic beverages trigger migraine attacks and activation of trigeminal neurons plays a role in migraine. We have investigated in guinea pigs whether ethanol by TRPV1 stimulation causes neurogenic inflammation in the trigeminovascular system. Ethanolevoked release of neuropeptides from slices of dura mater was abolished by Ca2+ removal, capsaicin pretreatment and the TRPV1 antagonist, capsazepine. Intragastric ethanol increased plasma extravasation in dura mater, an effect abolished by capsazepine and the NK1 receptor antagonist, SR140333, and caused vasodilation around the middle meningeal artery, an effect abolished by capsazepine and the CGRP receptor antagonist, BIBN4096BS. Vasodilation of meningeal vessels by TRPV1 activation and CGRP release may be relevant to the mechanism by which alcohol ingestion triggers migraine attacks.
Introduction
It is well accepted that migraine attacks may initiate spontaneously or may be triggered by specific factors, which, acting alone or in combination, enhance the probability of the attack (1). Observational studies have identified a variety of potential triggers, including fatigue (2), stress, mental tension (3, 4), weather changes (5, 6), menstruation (4, 6) and certain foods (6). Among nutritional factors, alcoholic beverages are well-known inducers of headache in healthy subjects (7). Migraineurs seem to be particularly sensitive to all alcoholic drinks, including red wine and beer (4, 8). The observed negative association between migraine and chronic alcohol consumption indirectly confirms the migraine-precipitating properties of alcohol (9). At present, the mechanism of alcohol-evoked headache is poorly understood. For example, sulphites or other non-alcoholic ingredients have been proposed to contribute to the migraine-precipitating effect of wine (10, 11). However, the possibility that ethanol (EtOH) per se triggers the migraine attack by a vasodilating effect has long been cited (12).
A subpopulation of primary nociceptive neurons, uniquely sensitive to capsaicin, the hot principle present in the genus Capsicum, contains and releases the neuropeptides calcitonin gene-related peptide (CGRP) and the tachykinins, substance P (SP) and neurokinin A (13, 14). CGRP and tachykinins, released from peripheral endings of primary sensory neurons, cause a series of biological responses collectively referred to as neurogenic inflammation (15). The CGRP-mediated arterial vasodilation, and tachykinin-mediated plasma protein extravasation, represent the main aspects of neurogenic inflammation (15). The release of sensory neuropeptide from terminals of trigeminal neurons distributed around meningeal blood vessels and their biological effects on these vessels have been proposed as the underlying mechanism of the migraine attack (16, 17). However, tachykinin NK1 receptor antagonists, despite their ability to block neurogenic plasma leakage, have failed to ameliorate migraine attacks in various clinical studies (18, 19). Increased CGRP levels in the blood of cranial origin during spontaneous or provoked migraine attacks (20, 21) have suggested a possible role of CGRP in migraine pathogenesis. The observation that CGRP administration causes migraine headache (22) and, more importantly, the clinical finding that BIBN4096BS, a high-affinity and selective CGRP receptor antagonist (23), reduces the pain and other symptoms associated with migraine attacks (24), indicate a primary role for neurogenic vasodilation and CGRP in migraine.
Sensitivity of nociceptive primary sensory neurons to capsaicin is given by the expression of the transient receptor potential vanilloid 1 (TRPV1) channel (25), which is also activated by noxious temperature (43–52°C), extracellular acidosis (pH 6–5) (26, 27), and diverse lipid derivatives (28–30). We have recently reported that EtOH stimulates TRPV1 channels and releases SP/CGRP from central and peripheral terminals of primary sensory neurons, by lowering the threshold temperature for channel activation (31). More recently, it has been shown that EtOH by a TRPV1-mediated neurogenic mechanism produces a CGRP-dependent vasodilation in porcine coronary and human gastric arteries (32). Finally, expression of TRPV1 mRNA and protein in human trigeminal ganglion neurons (33) supports the hypothesis that TRPV1-dependent release of CGRP contributes to the pain originating from the trigeminovascular system in man. The aim of the present study was to explore whether EtOH, by TRPV1 stimulation, activates trigeminal primary sensory neurons and by this mechanism releases sensory neuropeptides and causes neurogenic inflammatory responses, including arterial vasodilation in the guinea pig dura mater.
Methods
Animals and reagents
Male albino Dunkin Hartley guinea pigs (250–350 g; Charles River, Lecco, Italy) were acclimatized in cages (24 ± 0.5°C) for 1 week after delivery and were allowed free access to water and standard rodent diet. All experiments complied with European Community guidelines for animal care (86/609/EC). Formal approval to conduct the experiments described was obtained from the animal subjects review board of the University of Florence.
CGRP- and SP-like immunoreactivity release
Guinea pigs were terminally anaesthetized and decapitated. A trigeminal rhizotomy was performed via a ventromedian skull base craniotomy and the dura mater was carefully detached from the skullcap, at 4°C. Thick slices (∼0.3 mm) were placed in 2-ml chambers and superfused at 0.4 ml/min with a Krebs' solution of the following composition (in m
Plasma extravasation
Overnight fasted guinea pigs were anaesthetized intraperitoneally with sodium pentobarbital (60 mg/kg) and a plastic cannula was inserted into the trachea following tracheotomy to facilitate respiration. Guinea pigs were then artificially ventilated (60 breaths/min) through a rodent ventilator (7025; Ugo Basile, Comerio, Italy). Evans Blue dye (30 mg/kg) was injected into the jugular vein 1 min prior to the intraperitoneal (i.p.; 1 ml/kg) or intragastric (i.g.; 1 ml/kg) administration of EtOH or 1 min prior to the intravenous (i.v.) administration of SP (1 nmol/kg). After 30 additional minutes, the chest was opened and an incision made in the left ventricle. A cannula was then inserted through the left ventricle into the ascending aorta and approximately 200 ml of sterile saline (0.9%) was perfused at a pressure of 100 mmHg. The dura mater was then carefully removed, weighed and incubated in 1 ml of formamide for 24 h in the dark, at room temperature. The amount of extravasated Evans Blue was measured spectrophotometrically at 620 nm. Systemic pretreatments with capsazepine (4 mg/kg, i.p), SR140333 (1 mg/kg, i.v.), BIBN4960BS (1 mg/kg, i.v.) or their respective vehicles were given 10 min prior to injection of the dye.
Dural blood flow
Guinea pigs were fasted overnight and anaesthetized with an initial dose of urethane (1500 mg/kg, i.p.), followed by additional doses of 15 mg/kg, i.p., when required, to maintain the appropriate level of anaesthesia. Body temperature of the animals was kept at 37°C with a heating pad. The head of the animals was fixed in a stereotaxic frame. The scalp was incised along the midline and the parietal skin pulled aside. Using a manual drill (Harvard Instrument, Holliston, MA, USA), a cranial window of 4 × 6 mm was made into the parietal bone to expose the dura mater for the measurement of dural blood flow. At a distance of 1 mm from the meningeal layers, the probe (needle type, tip diameter 0.8 mm) of a Laser Doppler Flow Meter (Perimed Instruments, Milan, Italy) was fixed pointing near a branch of the middle meningeal artery. To minimize flow signals from the cortical blood vessels, recording sites were selected along the larger branches of the medial meningeal artery lying distant from the visible cortical blood vessels. Under these conditions, the registered laser Doppler signal derived nearly exclusively from the meningeal blood flow. The cranial window was carefully filled with a modified synthetic interstitial fluid (SIF) containing (m
Drugs
Drugs and reagents were obtained from the indicated companies: capsaicin, capsazepine, substance P, urethane and pentobarbital and Evans Blue dye (Sigma, Milan, Italy), SR140333 and BIBN4960BS were kind gifts of Dr X. Emonds-Alt (Sanofi Recherché, Montpellier, France), and Dr H. Doods (Boeringher-Mannheim, Mannheim, Germany), respectively.
Statistical analysis
All values are mean ± standard error of the mean (SEM). Differences between the groups were analysed by analysis of variance (
Results
CGRP and SP release
EtOH (3%) produced an increase in CGRP-LI and SP-LI outflow from slices of guinea pig trigeminal ganglia (195 ± 53 fmol/g per 20 min and 33 ± 8 fmol/g per 20 min, respectively, n = 6 each), which was reduced in both cases by >80% in experiments performed in a Ca2+-free medium, plus 1 m

Outflow of calcitonin gene-related peptide (CGRP)- and substance P (SP)-like immunoreactivity (LI) induced by ethanol from slices of guinea pig trigeminal ganglia (a,b) or dura mater (c,d) is reduced in a Ca2+-free medium, plus 1 m
Plasma extravasation
The baseline level of the extravasated Evans Blue dye in the guinea pig dura mater after the i.p. administration of EtOH vehicle (0.9% saline) was 15 ± 2 ng/g of tissue (n = 12). In a first set of experiments, i.p. administration of 1 ml/kg of EtOH increased significantly the amount ofextravasated Evans Blue dye in the dura mater (34 ± 2, n = 6, P < 0.05) (Fig. 2). Evans Blue dye extravasation induced by i.p. EtOH was practically abolished after pretreatment with SR140333 (1 mg/kg, i.v.) or capsazepine (4 mg/kg, i.p.) (Fig. 2a). In the second set of experiments EtOH (1 ml/kg) was administered intragastrically through a cannula that reached the gastric cavity. By this route of administration, Evans Blue dye extravasation evoked by EtOH vehicle (1 ml, 0.9% saline) was 17 ± 5 ng/g of tissue (n = 6). I.g. administration of EtOH increased significantly the Evans Blue dye extravasation (32 ± 2 ng/g of tissue, n = 6, P < 0.05). Extravasation induced by i.g. EtOH was abated after pretreatment with SR140333 (1 mg/kg, i.v.) or capsazepine (4 mg/kg, i.p.) (Fig. 2b). Pretreatment with SR140333 (1 mg/kg, i.v.), but not with capsazepine (4 mg/kg, i.p.) abolished Evans Blue dye extravasation evoked by SP (10 nmol/kg i.v.), indicating selectivity of capsazepine in EtOH-evoked extravasation (Fig. 2b). BIBN4960BS (1 mg/kg, i.v.) did not affect baseline Evans Blue extravasation (not shown). The increase in Evans Blue extravasation induced by i.g. EtOH (1 ml/kg) was not altered in animals pretreated with BIBN4960BS (1 mg/kg, i.v., 26.7 ± 2.1 ng/mg of tissue, n = 6) when compared with that observed in vehicle-treated animals (29.4 ± 3.2 ng/mg of tissue, n = 7).

(a) Plasma protein extravasation in the guinea pig dura mater induced by intraperitoneal (1 ml/kg, i.p.) or intragastric (1 ml/kg, i.g.) administration of ethanol was abated by pretreatment with the tachykinin NK1 receptor antagonist, SR140333 (1 mg/kg, i.v.) and by the TRPV1 antagonist, capsazepine (4 mg/kg, i.p., CPZ). (b) Effect of capsazepine on substance P (SP)-induced Evans Blue dye extravasation. Each entry is the mean ± SEM of at least six experiments. Sal (0.9% saline), ethanol vehicle; Veh, combination of capsazepine and SR140333 vehicles. ∗P < 0.05 vs. Sal; #P < 0.05 vs. Veh.
Dural blood flow
In preliminary experiments, basal blood flow in the dura mater of the guinea pig (259.0 ± 34.5 perfusion units, n = 5) was not affected by the local application of SIF (100 µl). In contrast, acetylcholine (1 m
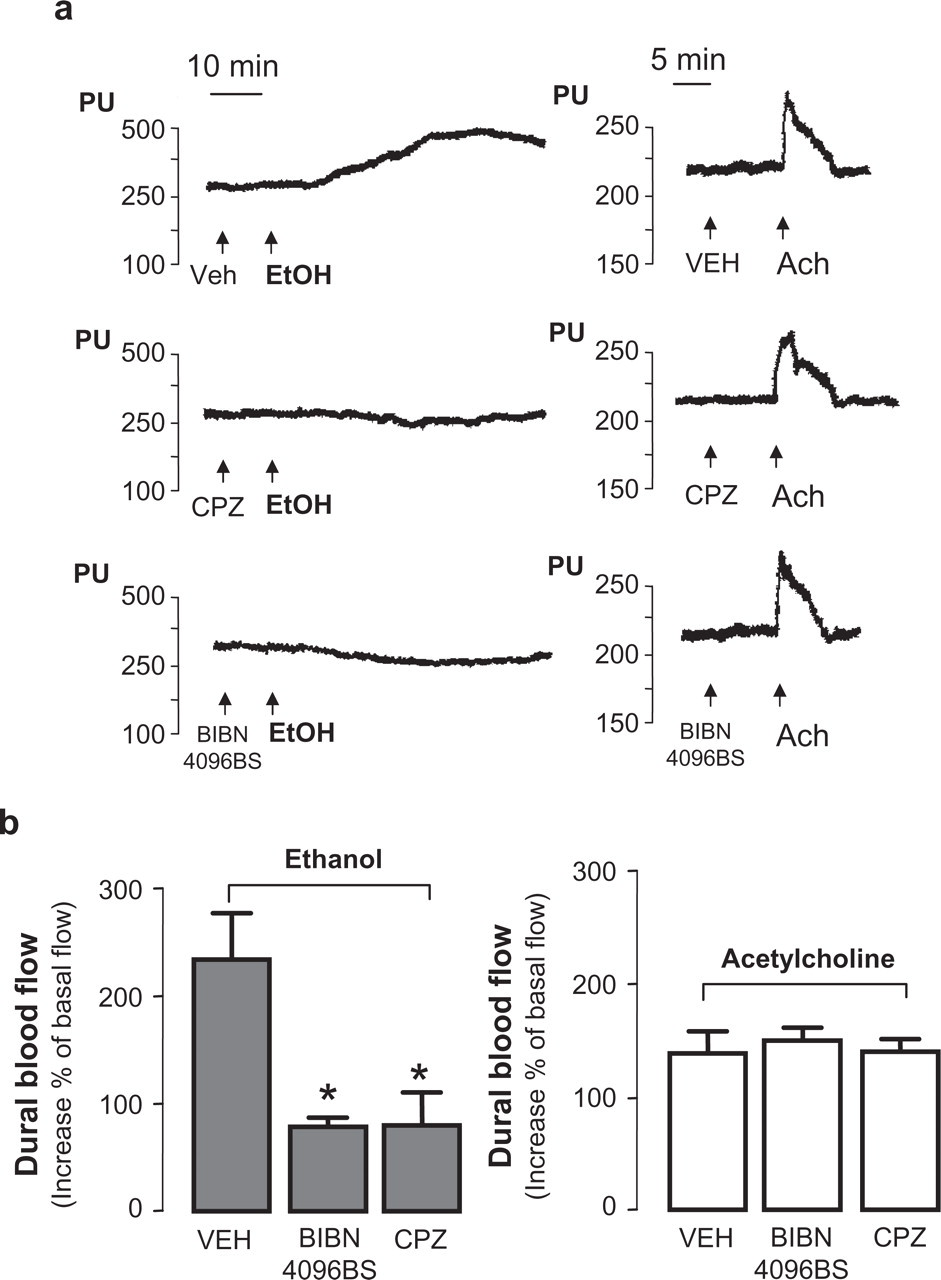
Typical tracings (a) and pooled data (b) of the effect of ethanol (1 ml/kg i.g., left side) and acetylcholine (Ach, 1 m
Discussion
This study provides evidence that EtOH causes neurogenic inflammatory responses, including plasma extravasation and arterial vasodilation, in the guinea pig meninges by a TRPV1-mediated release of sensory neuropeptides. Among many other neuronal effects, EtOH has been shown to stimulate nociceptive primary sensory neurons by lowering the threshold temperature for TRPV1 activation (31). This effect results in a Ca2+-dependent release of sensory neuropeptides in the spinal cord and in peripheral tissues, including the oesophagus (31) and airways (36). Our present experiments have shown that a similar phenomenon also occurs at the trigeminal level. In fact, EtOH increased the outflow of SP and CGRP from guinea pig trigeminal ganglia and from the dura mater, which is densely innervated by terminals of trigeminal neurons. Ca2+ dependency and sensitivity to capsaicin desensitization indicated that EtOH promotes a neurosecretory process from capsaicin-sensitive sensory cell bodies (trigeminal ganglion) or nerve terminals (dura mater). Finally, abolition by capsazepine confirmed also at the trigeminal level that EtOH-evoked release of sensory neuropeptides is mediated by activation of TRPV1. It is noteworthy that TRPV1 is coexpressed in human trigeminal ganglion neurons with the sensory neuropeptides, SP and CGRP (33). Thus, one may hypothesize that the mechanism described in the guinea pig may also occur in man.
EtOH-evoked release of sensory neuropeptides has been associated with neurogenic inflammatory responses, namely plasma extravasation in the rat oesophagus and in the guinea pig airways and bronchoconstriction in the guinea pig (31, 36). In this context, it is important to mention that plasma protein extravasation is a phenomenon exclusively mediated by activation of tachykinin NK1 receptors in postcapillary venules. Accordingly, EtOH-evoked plasma extravasation in the oesophagus and airways was abated by an NK1 receptor antagonist (31, 36). More recently, it has been shown that exposure to EtOH of pig coronary arteries or human gastroepiploic arteries is associated with a Ca2+-dependent and capsaicin-sensitive release of CGRP and with a CGRP-mediated arterial vasodilation (32). Likewise, plasma leakage, arterial vasodilation evoked by EtOH was inhibited by capsazepine. These in vivo findings confirm the conclusion derived from release experiments regarding the role of TRPV1 in EtOH-evoked sensory nerve stimulation. The ability of SR140333 to block plasma protein leakage in the dura mater shows that this effect is mediated by SP release and NK1 receptor activation on endothelial cells of postcapillary venules. There is strong evidence supporting the view that capsaicin-induced hyperaemia in mammals and in man is mediated by CGRP release from sensory nerve terminals (37). CGRP is one of the most potent vasodilator substances described to date (38), and CGRP receptor antagonists have been reported to inhibit neurogenic arterial vasodilation in a large series of in vivo preparations (39). However, it should be noted that the CGRP receptor antagonist, BIBN4096BS, did not affect cerebral or systemic circulation in humans (40). In addition, BIBN4096BS selectively prevented the vasodilation induced by CGRP and by transcranial electrical stimulation in the rat middle meningeal artery, but not in cortical pial arteries (41). This unique pharmacodynamic profile might result from the absence of a vasodilatory action of CGRP in the resting state and by the hydrophilic nature of this compound (23), which is unlikely to pass the blood–brain barrier (40, 41) Here, we have shown that BIBN4960BS and capsazepine inhibit EtOH-evoked increase in blood flow in the middle meningeal artery. Selectivity of these pharmacological interventions was supported by the observation that both capsazepine and BIBN4960BS did not affect acetylcholine-induced vasodilation. Thus, ingestion of EtOH causes neurogenic vasodilation in meningeal arterial vessels through TRPV1 stimulation and the release of CGRP from perivascular sensory nerve endings.
Neurogenic inflammation in the meninges has been proposed as the underlying mechanism of migraine headache (16). Originally, the term neurogenic inflammation was mainly, if not exclusively, referred to neurogenic plasma protein extravasation. The inhibitory action of antimigraine drugs, such as ergot derivatives and triptans, on neurogenic plasma protein leakage in the meninges further supported this view (42–44). However, clinical observation that selective and potent NK1 receptor antagonists (18, 19) did not ameliorate the pain and other symptoms of the migraine attack excluded a major role for tachykinins and NK1 receptors in migraine. At the same time, the hypothesis that the release of CGRP, which dilates cranial blood vessels (45) and stimulates sensory nerve transmission (46), key role in the mechanism of migraine has received much attention and experimental support. In addition to findings obtained in experimental animals, a recent clinical study has shown a beneficial effect of the high-affinity, peptoid antagonist of the CGRP receptor, BIBN4096BS (23), on the pain and other symptoms associated with the migraine attack (24).
EtOH may produce both vasoconstriction (47–49) and vasodilation (50, 51) on arterial vessels, including cerebral arteries. The different effect appears to be related, among several factors, to the size of the vessel and to species differences. However, the action of EtOH on sensory nerve terminals exclusively produces a vasodilatory effect, mediated by TRPV1 activation and CGRP release (32). This neuronal effect of EtOH may be relevant in clinical medicine, as EtOH-induced neurogenic vasodilation of coronary arteries (32) could contribute to the so-called French paradox (52). The protective effect of alcoholic beverages in coronary artery disease seems, in fact, directly dependent on their alcoholic content (53). However, alcoholic drinks, although protecting the heart, may be detrimental in migraineurs because they can trigger migraine attacks (8). This effect of alcohol has been linked to the vasodilatory properties of the substance (12). However, the precise mechanism through which EtOH provokes the migraine attack remains unknown. Here, we found that in guinea pigs, EtOH, by TRPV1 stimulation, produces, the excitation of primary sensory neurons, the release of sensory neuropeptides and the production of neurogenic inflammatory responses. The prolonged vasodilation, resulting from neural stimulation, observed following i.g. EtOH administration, somehow mirrors the delayed onset of the migraine headache after the ingestion of alcoholic beverages. Thus, both the excitatory action on nociceptive trigeminal nerve endings and the ability to cause CGRP-dependent vasodilation might contribute to trigger the migraine attack. The quantity of i.g. EtOH able to produce neuronal activation and neurogenic vasodilation (1 ml/kg) in guinea pigs corresponds to the alcoholic content of three to four glasses of wine, an amount which, in common experience, is sufficient to trigger a migraine attack. We propose that the ability of alcoholic beverages to trigger migraine attacks in susceptible individuals is due, at least in part, to the stimulation by EtOH of TRPV1 on trigeminal neurons and the resulting CGRP-mediated arterial vasodilation.
Acknowledgements
This study was supported by grants from MUR, Rome and Ente Cassa di Risparmio di Firenze, Florence, Italy.