
Abstract
The purpose of the study was to develop a mouse model to study trigeminovascular mechanisms using intravital microscopy on a closed cranial window. In addition, we studied exogenous and endogenous calcitonin gene-related peptide (CGRP)-mediated vasodilation in dural arteries. Arteries in C57BL/6Jico mice were constricted with endothelin-1, which reduced the baseline diameter by 65-75%. Subsequently, vasodilation was induced by α-CGRP, capsaicin or transcranial electrical stimulation of perivascular trigeminal nerves in the absence or presence of different concentrations of BIBN4096BS or sumatriptan. Both α-CGRP and capsaicin induced vasodilation in preconstricted arteries. Transcranial electrical stimulation also induced current-dependent relaxation of dural arteries with 100 μA producing maximal dilation in the control group. BIBN4096BS blocked the responses evoked by ä-CGRP and capsaicin, as well as electrical stimulation, whereas sumatriptan attenuated only vasodilation induced by electrical stimulation. This model is likely to prove useful in dissecting elements of the trigeminovascular system and for exploring pathophysiological aspects of migraine, especially in future studies using transgenic mice with mutations relevant to those observed in patients with migraine.
Introduction
Migraine is a common, chronic, episodic and disabling neurovascular disorder with largely unknown pathophysiology. It is widely believed that the headache phase of migraine involves distension of cranial blood vessels and activation of trigeminovascular nociceptive pathways (1, 2), or at least the perception of that activation (3). Electrical and chemical stimulation of the trigeminal innervation releases a range of vasoactive peptides, such as calcitonin gene-related peptide (CGRP) and substance P, resulting in dilation of these arteries. Based on this principle, the closed cranial window model in rats was developed by Williamson et al. (4). This innovative technique in rats involves thinning of parietal bone, thus allowing a continuous monitoring of meningeal blood vessels without opening the skull. Any changes in vessel diameter as a result of administration of various pharmacological agents or by transcranial electrical stimulation can be studied. This approach offers the possibility of directly studying various receptor systems implicated in modulating the trigeminovascular systemand thus their potential role in migraine. The use of intravital microscopy in rats has led to a better understanding of the underlying mechanism of trigeminal activation (4–6) and dural vasodilation (7–9), which are hallmarks of migraine pathophysiology. The current experimental setup of intravital microscopy uses wild-type animals (e.g. rats), whereas more advanced animal models, preferably with a genetic predisposition to disease, would be preferred in view of recent advances in genetic research in migraine (10). Until now, three migraine genes have been identified in families with familial hemiplegic migraine (FHM), a rare autosomal dominant subtype of migraine with aura (11–13). FHM genes CACNA1A, ATP1A2 and SCN1A are all involved in ion transport and encode subunits of Cav2.1(α1A) voltage-dependent P/Q type calcium channels, Na+/K+ pumps, or neuronal voltage-gated Nav1.1 sodium channels, respectively (11–13). However, the importance of the FHM genes in common migraine still needs validation.
The identification of migraine gene mutations in patients provides unique opportunities to generate transgenic mice with specific pathogenic mutations (e.g. knock-in mice), and thus a genetic predisposition to the disease. The recently described transgenic knock-in mouse model expressing the human R192Q pure FHM-1 mutation showed pure gain of functional effects (e.g. increased calcium influx through mutant calcium channels, increased neurotransmitter release and a reduced threshold for cortical spreading depression, the underlying cause of the migraine aura) and appears a promising model to study migraine pathophysiology (14).
Using these mutant mice in vivo in an intravital experimental set-up clearly seems a step forward in understanding the role of these mutations in the pathogenesis of migraine, but requires modification of the rat intravital microscopy model to the other species. Therefore, to develop and standardize this technique we studied the role of exogenous and endogenous CGRP-induced vasodilation in wild-type mice. CGRP is a potent vasodilator of dural arteries and it is well established that levels of this neuropeptide are increased during migraine (15, 16). Moreover, the role of CGRP in migraine pathogenesis (17, 18) is further strengthened by the fact that infusion of CGRP induces headache or migraine attacks in humans (19). In the present study, we used BIBN4096BS, a potent and selective CGRP receptor antagonist (20–22), which has been shown to be effective in aborting acute migraine attacks in a Phase II study (23). We also included in this study the conventional antimigraine drug sumatriptan, a 5-HT1B/1D receptor agonist (24).
Methods and materials
Surgical preparations
Male C57BL/6Jico mice (Charles River Laboratories, St Germain sur l'Abresle, France; 20–30 g) were anaesthetized throughout the experiment using pentobarbital sodium (80 mg/kg, i.p. and then 20 mg/kg/h, i.p.). The trachea was cannulated and connected to a pressure ventilator (small animal ventilator SAR-830 series; CWE, Inc., Ardmore, PA, USA). The jugular vein was cannulated for intravenous administration of drugs and the femoral artery was cannulated for continuous monitoring of blood pressure. Throughout surgery and in subsequent experiments, the core temperature of the animals was monitored via a rectal thermometer and maintained between 36.5 and 37.5°C by a blanket (homeothermic blanket system for rodents; Harvard Instruments, Edenbridge, UK). Subsequently, the mouse was placed in a stereotaxic frame; the skull was drilled thin until the dural arteries were clearly visible. As the mouse skull is very thin, care was taken to drill with a constant application of ice-cold saline. In about 15% of the mice, bleeding was observed underneath the skull, making visualization of the artery difficult; such animals were excluded from the study.
Intravital microscopy
The drilled area was covered with mineral oil to prevent drying of the skull and facilitate the visualization of the artery. The artery was captured with an intravital microscope (Leica MZ 16; Leica Microsystem Ltd, Heerbrugg, Switzerland) using a cyan filter on a cold source of light. A zoom lens (80–450× magnification) and a camera were used to capture the image of the dural artery, which was displayed on a standard television monitor. The dural blood vessel diameter was continuously measured using a video dimension analyser (Living Systems Instrumentation, Inc., Burlington, VT, USA) and data were displayed, recorded and measured using the WINDAQ data acquisition system (version 2.54; DataQ Instruments Inc., Akron, OH, USA).
Preconstriction of arteries
Unlike in rats, but similar to previous observations in guinea pigs (6), mice dural arteries were first constricted using endothelin-1 (ET-1, 7.5–10 µg/kg i.v.) before any pharmacological or electrical intervention, as it was not possible to observe significant vasodilation in arteries without preconstriction of the arteries. ET-1 was administered to induce a rapid vasoconstriction of around 65–75% in the blood vessel.
Effect of α-CGRP, capsaicin, BIBN4096BS and sumatriptan
The effect of α-CGRP (10 µg/kg) was studied in the presence or absence of the CGRP receptor antagonist BIBN4096BS (10, 30 and 100 µg/kg) or sumatriptan (3 mg/kg), whereas the effect of capsaicin (30 µg/kg) was studied in the absence or presence of BIBN4096BS (100 µg/kg). Five minutes before the administration of ET-1, vehicle (control experiments), BIBN4096BS or sumatriptan were administered as an i.v. bolus. Two minutes after the administration of ET-1, α-CGRP or capsaicin were administered i.v. as a fast bolus or slowly in 30 s, respectively. The arterial diameter was recorded for another 5 min.
Transcranial electrical stimulation and the influence of BIBN4096BS and sumatriptan on neurogenic vasodilation
In the preparations where transcranial electrical stimulation was used to evoke dilation of the dural blood vessels, a bipolar stimulating electrode (NE 200X; Clark Electromedical, Edenbridge, UK) was placed on the surface of the cranial window within approximately 200 µm of the vessel of interest. The surface of the cranial window was stimulated at 5 Hz, 1 ms for 10 s (Stimulator model S88; Grass Instruments, West Warwick, RI, USA). For neurogenic dural vasodilation, we initially started with a current intensity (monitored on an oscilloscope, model 54601A; Hewlett Packard, Palo Alto, CA, USA) of 50 µA and increasing with 50 µA steps until a maximal level of dilation was achieved, usually at 150 µA. In view of the fact that the contractile response to ET-1 began to fade off after about 7 min, transcranial stimulations were performed at intervals of 1.5 min, starting 2 min after the administration of ET-1. We also investigated the effect of pretreatment of BIBN4096BS (30 or 100 µg/kg) or sumatriptan (3 mg/kg) on the vasodilation induced by transcranial electrical stimulation. Both BIBN4096BS and sumatriptan were administered 5 min before the administration of ET-1.
Data analysis
The dural vessel diameter has been expressed in arbitrary units (AU) and reported as mean ± SEM. Diameter measurements were made in AU, because the experimental set-up changed depending on the portion of the artery segment studied and magnification of the camera to get the best image to monitor the diameter. The changes in the mean arterial blood pressure were expressed as mmHg. Within each group,
Compounds
The compounds used in the present study (obtained from the sources indicated) were: human α-CGRP and human ET-1 (both from NeoMPS S.A., Strasbourg, France), BIBN4096BS (gift: Dr H. Doods, Boehringer Ingelheim Pharma K.G., Biberach, Germany), sumatriptan succinate (gift: GlaxoSmithKline, Stevenage, UK) and capsaicin (Sigma Chemical Co., Steinheim, Germany). α-CGRP, ET-1 and sumatriptan were dissolved in water. Capsaicin (1 mg/ml) was dissolved in a mixture of Tween-80, ethanol 70% and water (1 : 1 : 8). BIBN4096BS was dissolved in 4% HCl (1 N) to obtain a 0.01 M stock solution, which was further diluted with distilled water. All stock solutions were stored at −80°C until required. Just before use, the stock solutions were further diluted to appropriate concentrations in isotonic saline for injection.
Results
Representative recordings of vessel diameter
Figure 1 depicts original dural arterial images obtained from two mice treated with vehicle or BIBN4096BS (100 µg/kg). Using the video dimension analyser, we found that the diameter of the dural arteries that we studied varied between 20 and 30 µm in the non-constricted state (baseline). In both mice, ET-1 (10 µg/kg) elicited a clear reduction of vessel diameter and CGRP (10 µg/kg) reversed the constrictor effect of ET-1 in the animal treated with vehicle. The vasodilator response to CGRP was not observed in the mouse treated with BIBN4096BS. Also, it may be noted that the cerebral vessel (Fig. 1, top panels) did not respond to ET-1 or CGRP.

Representative video-microscopic images of dural arteries (DA) in two mice treated with vehicle (top panels) or 100 µg/kg BIBN4096BS (bottom panels). Left panels, 5 min after administration of vehicle or BIBN4096BS (baseline); middle panels, 2 min after the administration of endothelin-1 (ET-1) (10 µg/kg); right panels, 2 min after the administration of calcitonin gene-related peptide (CGRP) (10 µg/kg). The white line in the middle of each panel tracks vessel diameter. Note that ET-1 constricts dural arteries in both mice, while CGRP induces vasodilation in the vehicle-treated, but not in BIBN4096BS-treated mouse. The cerebral vessel (CV) to the right of the dural artery in the top panels does not respond to ET-1 or CGRP.
Endothelin-1-induced dural artery constriction
Pilot experiments revealed that doses of ET-1 <7.5 µg/kg produced variable and unstable constriction of dural arteries, making the experimental window unpredictable. ET-1 in doses of 7.5–10 µg/kg induced 65–75% reduction in the baseline diameter. Higher doses of ET-1 (15–20 µg/kg) caused a total collapse of the artery and these arteries did not dilate sufficiently in response to α-CGRP, even in the absence of antagonists. Doses of ET-1 (7.5–10 µg/kg) provided a stable baseline up to 7 min, which was sufficient to perform the protocols for the current study.
ET-1 significantly reduced the baseline diameter from 60.9 ± 4.8 to 14.3 ± 2.6 AU 2 min after administration (overall significance across the cohort: F 3,26 = 6.88, P < 0.0015, n = 8). The diameter of vessels did not change significantly up to 7 min (23.1 ± 3.0 AU), but after 10 min the diameter significantly increased to 32.3 ± 2.4 AU compared with the vessel diameter 2 min after the administration of ET-1 (Fig. 2). The percentage decreases in diameter by ET-1 compared with baseline were 76%, 74%, 64% and 48% after 2, 4, 7 and 10 min, respectively. Also, the mean arterial blood pressure compared with the baseline value (67 ± 8 mmHg) increased significantly to 70 ± 6, 61 ± 4, 56 ± 4 and 48 ± 5 mmHg at 2, 4, 7 and 10 min after administration of ET-1, respectively.

Effect of endothelin-1 (ET-1) on dural artery diameter at different time points after its administration. ∗Significant difference vs. the baseline diameter (P < 0.05); †significant difference vs. diameter 2 min after ET-1 (P < 0.05).
Responses to CGRP
A bolus injection of α-CGRP (10 µg/kg) induced dural artery dilation (overall significance across the cohort: F 2,63 = 55.93, P < 0.0001, n = 22). There was a significant reduction in dural blood vessel diameter after ET-1 (7.5–10 µg/kg) injection (Table 1). Two minutes after α-CGRP injection, there was a significant increase in dural blood vessel diameter to 44.6 ± 2.8 AU (P < 0.01). After 5 min the dural blood artery diameter had significantly increased further to 57.0 ± 2.5 AU (P < 0.01, Fig. 3) compared with the diameter 2 min after ET-1 administration. α-CGRP decreased the mean arterial blood pressure from 111 ± 10 mmHg to 42 ± 8 and 48 ± 11 mmHg at 2 and 5 min post administration, respectively.

Effect of BIBN4096BS (a) and sumatriptan (b) on α-calcitonin gene-related peptide (CGRP)-induced dilation of dural artery preconstricted with endothelin-1 (ET-1) (7.5–10 µg/kg). †Significant difference vs. dural artery diameter after ET-1 (P < 0.05).
Dural artery vasodilator responses to α-calcitonin gene-related peptide (CGRP), capsaicin and transmural electrical stimulations in mice treated with vehicle, BIBN4096BS or sumatriptan
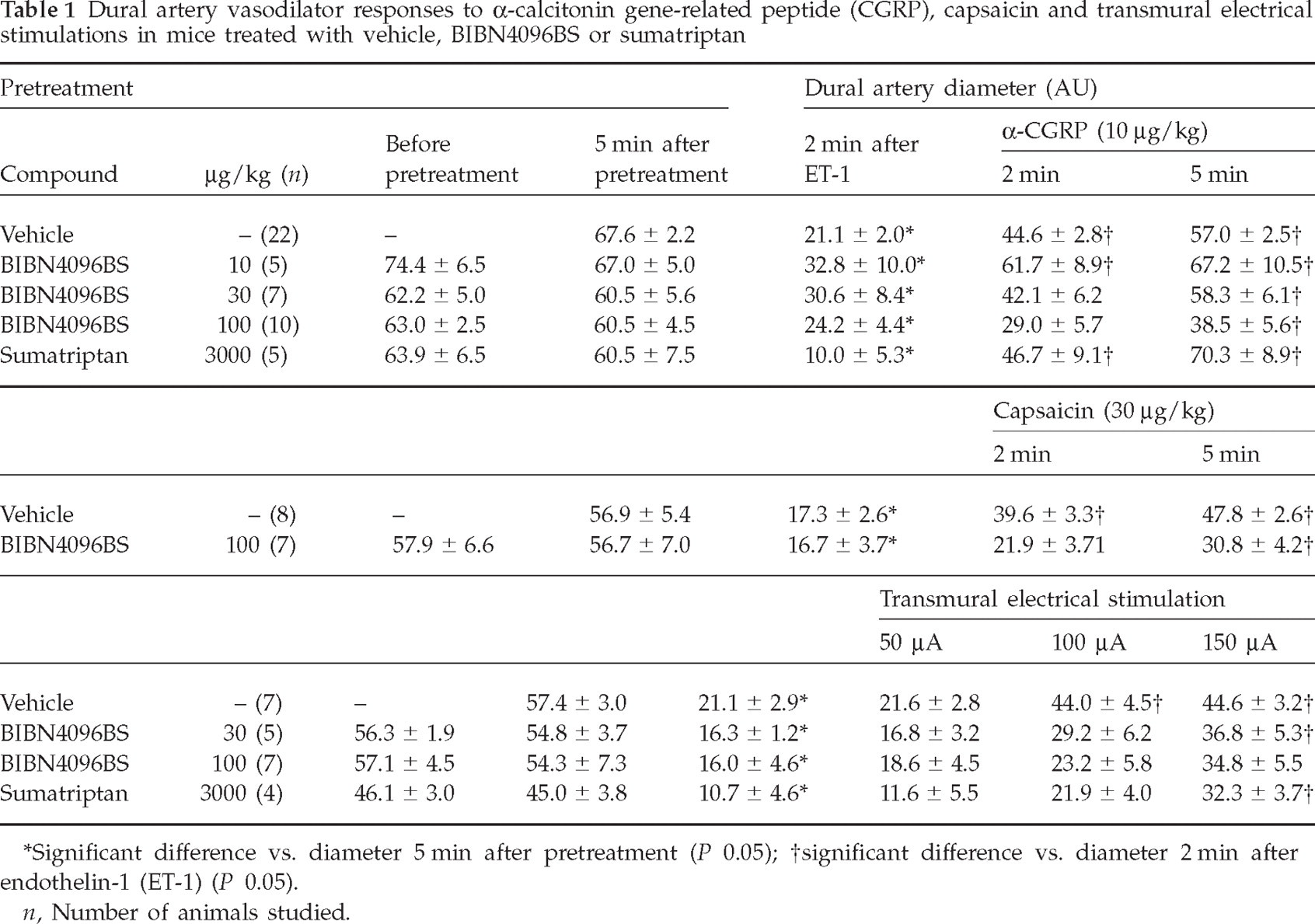
Significant difference vs. diameter 5 min after pretreatment (P < 0.05);
significant difference vs. diameter 2 min after endothelin-1 (ET-1) (P < 0.05).
n, Number of animals studied.
Pretreatment with BIBN4096BS (10, 30 and 100 µg/kg) did not cause any significant change per se in the dural artery diameter (Table 1) or mean arterial blood pressure (before, 78 ± 6 mmHg; after 100 µg/kg BIBN4096BS, 83 ± 6 mmHg). When a low dose of BIBN4096S (10 µg/kg) was administered, it did not antagonize α-CGRP-induced relaxations. However, higher doses of BIBN4096BS (30 and 100 µg/kg) produced a significant blockade of α-CGRP-induced vasodilation, as there was no increase in vessel diameter 2 min after CGRP administration; after 5 min the diameter increased significantly in both groups (Figs 1 and 3a, Table 1). The decrease in mean arterial blood pressure at the highest dose of BIBN4096BS was 26 ± 6 and 29 ± 4 mmHg at 2 and 5 min after administration of α-CGRP, respectively.
Administration of sumatriptan (3 mg/kg) neither changed the mean arterial blood pressure (60 ± 6 and 63 ± 5 mmHg before and after administration of sumatriptan, respectively), nor did it affect α-CGRP-induced relaxation, as there was a significant vasodilation after 2 min of administration, similar to the control group (Fig. 3b).
Responses to capsaicin
Capsaicin (30 µg/kg) was administered slowly during 30 s, 2 min after administration of ET-1. Capsaicin induced a significant increase in the vessel diameter across the cohort (F 2,21 = 31.1, P < 0.001) and after 2 min of administration arteries dilated significantly (Table 1). The vehicle of capsaicin did not affect the dural artery diameter or blood pressure (data not shown). After treatment with capsaicin the blood pressure decreased by 8 ± 4 and 20 ± 6 mmHg after 2 and 5 min, respectively. BIBN4096BS (100 µg/kg) pretreatment significantly blocked capsaicin-induced relaxation (Fig. 4).

Effect of BIBN4096BS on capsaicin-induced dilation of dural artery preconstricted with endothelin-1 (ET-1) (7.5–10 µg/kg). †Significant difference vs. dural artery diameter after ET-1 (P < 0.05).
Responses to transcranial electrical stimulation
Transcranial electrical stimulation induced neurogenic dural vasodilation; the arterial diameter increased with increasing current intensity across the cohort (overall significance across the cohort: F 3,21 = 7.64, P < 0.001). There was little change in diameter after a 50-µA current compared with the diameter after ET-1. After a 100-µA current, there was a significant increase in dural artery diameter compared with that after ET-1 (P < 0.01), but a current of 150 µA did not increase the vessel diameter further (Fig. 5, Table 1).

Effect of BIBN4096BS (a) and sumatriptan (b) on neurogenic vasodilation in the preconstricted dural artery. †Significant difference vs. dural artery diameter after endothelin-1 (ET-1) (P < 0.05).
When mice were pretreated with BIBN4096BS (30 µg/kg), there was no significant increase in vessel diameter after 100 µA current, although at a higher current intensity of 150 µA the increase was significant. At the highest dose of BIBN4096BS (100 µg/kg) there was no significant increase in vessel diameter even at 150 µA. Sumatriptan also significantly blocked the responses to transcranial electrical stimulation at 100 µA, although at the higher intensity of 150 µA there was a significant increase in the arterial diameter of the constricted blood vessels (Fig. 5, Table 1). There were no significant changes observed in mean arterial blood pressure after the electrical stimulation.
Discussion
This study demonstrates that intravital microscopy of a closed cranial window in mice is a reliable model to study the dural component of the trigeminovascular system. This is the first study in mice to measure directly the dural artery diameter in vivo.
Unlike in the rat closed cranial window model, but similar to what has previously been observed in guinea pigs (6), in the mouse model the meningeal arteries require preconstriction in order to observe subsequent vasodilation. We used ET-1 for preconstriction since it induced a rapid vasoconstriction and provided a stable experimental window for approximately 7 min. This is a relative disadvantage in comparison with the rat model, as it introduces an additional, but necessary, variable in the experimental conditions. α-CGRP-induced relaxation in the preconstricted arteries is in accordance with previous observations in an in vivo mouse skin vasculature assay (25). In this study, a relatively high dose of α-CGRP was used to induce dilation, probably because of the pretreatment with ET-1 to constrict the dural vessels. Further, it should be kept in mind that human α-CGRP is less potent in rodents (26) than in human blood vessels (27). BIBN4096BS did not induce vasoconstriction on its own, nor did it affect the mean arterial blood pressure, even at the highest dose used; this is in agreement with previous findings in rat (9, 28) and human in vivo studies (29, 30). α-CGRP-induced vasodilation was dose-dependently blocked by BIBN4096BS, in line with a recent intravital microscopy study in the rat (9). In our experiments, the 5-HT1B/1D receptor agonist sumatriptan did not block relaxation induced by exogenously administered α-CGRP. Similarly, rizatriptan also had no effect on the vasodilation in response to exogenous CGRP in the guinea pig (6). Furthermore, when administered 5 min prior to ET-1, sumatriptan did not induce any significant vasoconstriction, indicating that this 5-HT1B/1D receptor agonist is devoid of vasoconstrictor properties in the mouse meningeal artery. In the human middle meningeal artery in vitro sumatriptan exerts a potent vasoconstrictor effect (31), but it does not decrease the vessel diameter in rats (4). As sumatriptan is able to reach the dural vasculature in the mouse without hindrance of the blood–brain barrier, the discrepancy in responses to sumatriptan can probably be attributed to differences in distribution of 5-HT receptors in the cranial vasculature or receptor affinity.
Capsaicin, a vanilloid (TRPV1) receptor agonist, also dilated the preconstricted arteries in the present study. Capsaicin has been widely used in animal models to induce cranial vasodilation (32, 33) and it acts by releasing endogenous CGRP. Also in the present study, the capsaicin-induced relaxation was mainly mediated via the release of CGRP, as demonstrated by the blockade with the CGRP receptor antagonist, BIBN4096BS, at 2 min. The fact that the vasodilator response to capsaicin was not significantly inhibited by BIBN4096BS at 5 min could be caused by a further increase of the effect of capsaicin after 2 min, as observed in the control experiments. Thus, the dose of BIBN4096BS applied might not have been sufficiently high to block completely the higher response observed at 5 min. Further, besides CGRP, capsaicin may release other vasodilators, such as substance P (34). Finally, there is a tendency for the diameter to increase at the greatest time points, although the baseline diameter did not significantly change during the experimental time window that we used (see Fig. 2). Transcranial electrical stimulation of the closed cranial window induced current-dependent dilation of the meningeal arteries. Earlier studies on a closed cranial window in rats used the voltage that induced maximal dilation, while we employed a stepwise increase in voltage, yielding currents of 50, 100 and 150 µA. This improvement over the existing model provides the flexibility to study the meningeal vessels at increasing current intensities and thus differences in the threshold stimulation between various animals may be detected. Further, a stepwise increase in stimulus intensity allows a more detailed assessment of the effects of inhibitors of the response to transcranial electrical stimulation; for example, 30 µg/kg of BIBN4096BS inhibited the vasodilator response to a stimulus intensity of 100 µA, but not to the supramaximal stimulus obtained with 150 µA. In contrast, the higher dose of BIBN4096BS (100 µg/kg) inhibited vasodilator responses to both 100 µA and 150 µA. In view of the dose-dependent blockade by BIBN4096BS, it can be concluded that, similar to the rat and guinea pig models (6), endogenous release of CGRP from perivascular nerves is a major component mediating vasodilation induced by transcranial electrical stimulation. Indeed, a low stimulus intensity during a short period, as employed in the present study, has been reported to selectively activate Aδ-sensory fibres, predominantly releasing CGRP (4). Relatively high doses of BIBN4096BS (30 and 100 µg/kg) were required to inhibit vasodilation, in line with the reported lower potency of this CGRP antagonist in rodents (9, 22). In addition, as mentioned earlier regarding the blockade of vasodilator responses to capsaicin by BIBN4096BS, the inhibitory actions of BIBN4096BS and sumatriptan at the highest stimulus intensity (i.e. at later time points) may have been underestimated because of a non-significant decline of the vasoconstrictor response to ET-1. Other agents, such as nitric oxide, may also contribute to neurogenic vasodilation, as observed in rats and cats (35, 36) and, indeed, such a dilation would be resistant to BIBN4096BS blockade. After electrical stimulation as well as α-CGRP, the dural artery diameter did not recover completely; this might be due to persistence of the constrictor effect of ET-1 (Fig. 2). The other possibility is that the stimulus intensity and the dose of CGRP were not high enough to dilate the artery to the preconstriction level.
Sumatriptan blocked the responses to transcranial electrical stimulation at 100 µA, compared with the control group. This may be due to the action of sumatriptan on presynaptic 5-HT1B/1D receptors, blocking the exocytosis of endogenously stored neuropeptides, including CGRP, as sumatriptan per se did not cause any change in vessel diameter. Similarly, in the rat cranial window model, 5-HT1B/1D receptor agonists have been shown to block neurogenic vasodilation (4, 6).
In conclusion, this mouse model of a closed cranial window can be used to explore further the mechanism of action and pharmacology of existing and putative antimigraine drugs. The main advantage of the mouse compared with the rat model is that this model provides the potential to use genetically modified mice. This is especially relevant, because recent advances in migraine genetics made it possible to generate transgenic knock-in mice harbouring pathogenic mutations observed in migraine patients (10, 14). This mouse model may provide an important insight into the effect of these mutations on dural artery vasodilation and, thus, offers an additional avenue to investigate the pathophysiology of migraine.
Footnotes
Acknowledgements
The advice of Dr David J. Williamson (Merck Sharp & Dohme Research Laboratories, UK) is acknowledged with gratitude. We thank Mr Rob H. van Bremen (Department of Experimental Cardiology, Erasmus MC, the Netherlands) for his technical assistance.