
Abstract
Although the understanding of migraine pathophysiology is incomplete, it is now well accepted that this neurovascular syndrome is mainly due to a cranial vasodilation with activation of the trigeminal system. Several experimental migraine models, based on vascular and neuronal involvement, have been developed. Obviously, the migraine models do not entail all facets of this clinically heterogeneous disorder, but their contribution at several levels (molecular, in vitro, in vivo) has been crucial in the development of novel antimigraine drugs and in the understanding of migraine pathophysiology. One important vascular in vivo model, based on an assumption that migraine headache involves cranial vasodilation, determines porcine arteriovenous anastomotic blood flow. Other models utilize electrical stimulation of the trigeminal ganglion/nerve to study neurogenic dural inflammation, while the superior sagittal sinus stimulation model takes into account the transmission of trigeminal nociceptive input in the brainstem. More recently, the introduction of integrated models, namely electrical stimulation of the trigeminal ganglion or systemic administration of capsaicin, allows studying the activation of the trigeminal system and its effect on the cranial vasculature. Studies using in vitro models have contributed enormously during the preclinical stage to characterizing the receptors in cranial blood vessels and to studying the effects of several putative antimigraine agents. The aforementioned migraine models have advantages as well as some limitations. The present review is devoted to discussing various migraine models and their relevance to antimigraine therapy.
Keywords
Introduction
Migraine is a complex, disabling, multifactorial, typically episodic neurovascular disorder of unknown aetiology; it affects a significant proportion of the adult population and it is a familial syndrome with a genetic component (1–3). Migraine attacks are characterized by an intense, throbbing and pulsatile headache, which is often unilateral in onset and is accompanied by anorexia, nausea, vomiting, photophobia and/or phonophobia (3). Furthermore, one-third of migraine sufferers experience aura, a focal neurological symptom including scintillating scotoma, muscle weakness and sensory disturbances (2, 3).
The pathophysiology of migraine involves the activation of the trigeminovascular system which, in turn, results in headache produced due to changes in cranial blood vessels (2, 4). These pain-producing cranial blood vessels are innervated by trigeminal nerves that release calcitonin gene-related peptide (CGRP), which produces a potent vasodilation (5). This dilation activates trigeminal nerves, which transmit vascular nociception to higher centres in the central nervous system (5, 6).
On the basis of these observations, various experimental models for migraine have been developed (7, 8); these will be considered in this review in detail.
Pathophysiology of migraine
The knowledge of the underlying pathophysiology of migraine has enhanced over the years and it is now well accepted that migraine is principally a disorder of the brain (2). Based on clinical symptoms, the pathophysiology of migraine can be divided into three phases: (i) the trigger phase characterized by neuronal hyperexcitability, (ii) the aura phase, possibly involving cortical spreading depression and, finally, (iii) the headache phase due to cranial vasodilation precipitated by activation and sensitization of the trigeminal system at the peripheral and central levels (4, 9–11). Exploring each phase of migraine reveals unique mechanisms and divulges novel therapeutic targets.
Trigger phase
Several lines of evidence, including studies using positron emission tomography, indicate that brainstem structures, rather than cortical structures, are activated during a migraine attack (2, 9). This activation remained unaffected after the resolution of the headache by sumatriptan treatment, suggesting that brainstem activation is a fundamental part of migraine (2, 12). In addition, certain genetic abnormalities may be responsible for altering the response threshold to migraine-specific triggers in the brain, e.g. mutations of the P/Q-type calcium channel gene that plays an important role in familial hemiplegic migraine (1, 2). The subsequent events following the trigger phase leading to the symptoms observed during the aura and headache phases can be explained on the basis of neurovascular involvement (6, 11).
Aura phase
Up to 15–30% of migraine sufferers experience aura symptoms that last for 5–60 min before the onset of headache and may involve cortical spreading depression (13, 14). Cortical spreading depression is a wave of transient intense spike activity that spreads along the cortex slowly at rates of between 2 and 6 mm/min, possibly leading to long-lasting neuronal inhibition (2, 14). With the advent of non-invasive methods, including magnetic resonance imaging (MRI) and magnetoencephalography (15), it has been shown that a wave of cortical spreading depression is followed by a decrease in regional cerebral blood flow (2, 14). Most clinicians believe that migraine aura is due to neuronal dysfunction, probably resulting from cortical spreading depression (15, 16), rather than ischaemia.
Headache phase
Although elegant functional neuroimaging procedures have refined the underlying concept of migraine, the mechanism by which the aura changes into headache still remains elusive (13). Clinical and experimental considerations suggest that the pathogenesis of the migraine headache is intimately linked to the trigeminal innervation, which when activated, possibly following cortical spreading depression, causes dilation of cranial blood vessels, including arteriovenous anastomotic shunts (11, 17, 18). The involvement of shunt vessels in migraine has once again attracted attention because patients with right to left cardiac shunts have been reported to have a high incidence of migraine headaches that are substantially reduced after shunt repair (see 19–21). Moreover, it is well known that patients with cranial arteriovenous malformations have a high incidence of migraine, which is reduced after correction of such malformations (22–25). Thus, migraine pain is due to activation of the nociceptors in intracranial structures, in concert with a reduction in the function of endogenous pain-control pathways (2). This nociceptive information from the cranial blood vessels is conveyed to central neurons in the trigeminal sensory nucleus that, in turn, relay the pain signals to higher centres, where headache is perceived (4, 13). In addition, trigeminal pathways may become sensitized (10, 26, 27) as well as release CGRP, thus reinforcing vasodilation relaying the nociceptive impulses to the central nervous system (11).
Experimental models for migraine
In general terms, experimental models are a part of strategy that is employed to develop new and better therapeutic agents for a particular ailment (Fig. 1). Experimental models are developed on the basis of pathophysiological features of the disease and the pharmacological action of the existing drugs. The new compounds found active in the models are subjected to clinical evaluation. If the compound is devoid of clinical efficacy and pharmacokinetic reasons are excluded, the model must be modified or discarded. On the other hand, when the compound is found clinically effective, not only does it benefits patients, but further evaluation of its pharmacology leads to modification of experimental models and, in some cases, to entirely new models that may yield new compounds ultimately becoming novel therapeutic agents.

Experimental models based on disease pathophysiology are critical for the drug development process. New compounds found effective in these models undergo clinical evaluation. If found effective, the pharmacological properties of the new drugs help in evolving new experimental models, which provide the basis for improved new drugs.
The experimental models for migraine developed in the last two decades (11, 18) investigate principles such as: (i) constriction of dilated extracranial blood vessels, including carotid arteriovenous anastomoses (e.g. carotid vasculature or isolated blood vessels; vascular involvement); (ii) inhibition of the trigeminal system (e.g. blockade of plasma protein extravasation and/or central trigeminal inhibition, neurogenic involvement) (2); and (iii) a combination of the two (e.g. inhibition of neurogenic vasodilation).
Migraine models based on vascular involvement
In vivo animal models
Constriction of carotid arteriovenous anastomoses in anaesthetized pigs. Although a complete understanding of migraine pathogenesis remains elusive, there is little doubt that dilation of cranial blood vessels, including carotid arteriovenous anastomoses, is involved in the headache phase of migraine (11, 18). In addition to headache, migraine patients also experience facial paleness, reduction in facial temperature, an increase in temporal artery pulsations and swelling of the frontal vein on the side of the headache (28, 29). Thus, Heyck (17) measured the oxygen saturation difference between the arterial and external jugular venous blood samples (A–V SO2 difference) during and after the headache phase of migraine and compared it with the healthy control groups. As shown in Fig. 2 (left panel), he observed that the A–V SO2 difference was abnormally small during the headache phase of migraine, probably due to dilation of the carotid arteriovenous anastomoses, and that this decrease was normalized after spontaneous or drug-induced (ergotamine) alleviation of the headache (17).
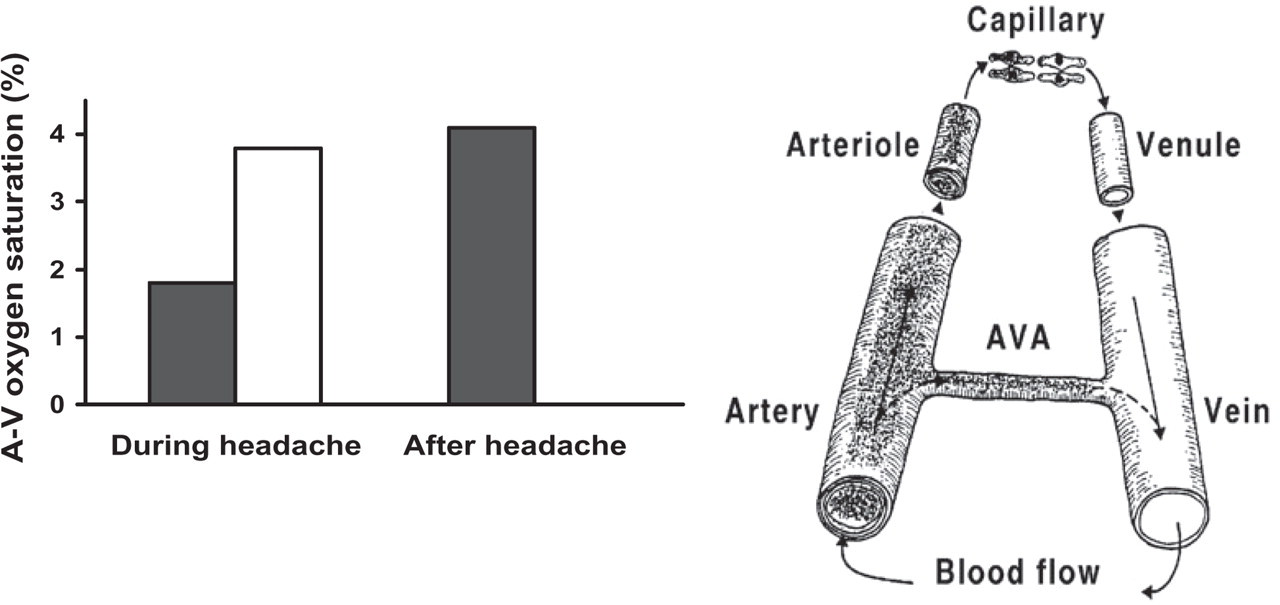
Arterio-jugular vein oxygen saturation difference on the ipsilateral side (▪) is lower than on the contralateral side (□) during migraine headache (left panel, data from Heyck (17)), probably due to opening up of arteriovenous anastomoses (AVA), which, being direct communications between arteries and vein, shunt oxygen-rich blood to the venous side (right panel).
Arteriovenous anastomoses are precapillary communications between the arteries and veins (Fig. 2, right panel); they are predominantly located in the head skin, ears, nasal mucosa, eyes and dura mater in several species, including humans and pigs (30). In conscious animals, the arteriovenous anastomoses are constricted under a strong influence of the sympathetic neuronal tone, thereby shunting only a small (<3%) fraction of the total carotid blood flow (31, 32). In contrast, under pentobarbital anaesthesia, approximately 80% of the total carotid blood flow in the pig is shunted via arteriovenous anastomoses into the jugular venous circulation (32). Consequently, opening of the carotid arteriovenous anastomoses during migraine shunts a large quantity of oxygenated blood directly into the veins, thereby resulting in facial pallor, a lowering of skin temperature and an increase in vascular pulsations (30). This increase in vascular pulsations stimulates the so-called ‘stretch receptors’ present in the wall of blood vessels, with ensuing activation of perivascular trigeminal nerves containing peptides (e.g. CGRP) (11, 18). The trigeminal cranial nerve conveys nociceptive information to central trigeminal nuclei that, in turn, relay the pain signals to higher centres where headache is perceived (5).
In accordance with the above findings, it is reasonable to assume that counteracting carotid arteriovenous anastomotic vasodilation may abort migraine (11, 18). Therefore, an animal experimental model in anaesthetized pigs was developed; radioactive microspheres were used to measure the carotid arteriovenous anastomotic blood flow and the effects of antimigraine drugs on this parameter (30). Over the years, this model has proven predictive of antimigraine activity in the clinic (11, 18) and, in fact, two of the early molecules in the sumatriptan development programme were evaluated and found active in this model (P.R. Saxena, personal observations, 1981). The major advantage of the porcine model is that one can simultaneously study different vascular beds in order to evaluate the cranioselectivity of antimigraine drugs (18). Based on this notion, all acutely active antimigraine agents, including ergot alkaloids, triptans as well as α-adrenoceptor agonists, potently constrict the porcine carotid bed and the corresponding arteriovenous anastomoses (18, 33–37); Fig. 3 shows that ergotamine, sumatriptan and phenylephrine dose-dependently decrease arteriovenous anastomotic blood flow. These results, together with Heyck’s (17) observations of decreased A–V SO2 difference during migraine headache, shed light on the mechanisms of action of acutely acting antimigraine drugs. Hence, the porcine carotid (arteriovenous anastomotic) circulation is an experimental model highly predictive of antimigraine activity, but this model will only pick up potential antimigraine drugs acting via vascular mechanisms.

Arteriovenous anastomotic (AVA) blood flow measured at baseline and after infusions of ergotamine (2.5, 5 and 10 μg/kg, i.v.), sumatriptan (30, 100 and 300 μg/kg, i.v.) and phenylephrine (1, 3 and 10 μg/kg per min, intracarotid artery infusion). All values are expressed as mean ± SEM. ∗P < 0.05 vs. baseline values. Data, presented as mean ± SEM, are from: ergotamine (33), sumatriptan (34) and phenylephrine (35).
Constriction of the canine external carotid bed. All acutely effective antimigraine drugs available thus far also produce selective constriction of cephalic blood vessels in the dog, including the external carotid artery, its temporal branches and arteriovenous anastomoses (11, 18). In this context, although serotonin (5-HT) (38), sumatriptan (38, 39), ergotamine and dihydroergotamine (40, 41) elicit a selective external carotid vasoconstriction in vagosympathectomized dogs, different mechanisms are involved. Thus, 5-HT and sumatriptan produced a dose-dependent vasoconstriction amenable to blockade by the 5-HT1B/1D receptor antagonist GR127935 (42), suggesting that 5-HT1B/1D receptors are involved, but the profile of antagonism is different. The vasoconstrictor responses to both 5-HT and sumatriptan are completely blocked by GR127935, but the in case of 5-HT a dose-dependent vasodilator component is unmasked (43).
A further pharmacological analysis of the 5-HT-induced vasodilator responses in animals pretreated with GR127935 revealed that this effect is mediated by the 5-HT7 receptor (44). Moreover, with the use of selective 5-HT1B (SB224289) and 5-HT1D (BRL15572) receptor antagonists, it was shown that the external carotid vasoconstrictor response is mediated by the 5-HT1B (not 5-HT1D) receptor (45).
The ergot alkaloids ergotamine and dihydroergotamine (46, 47) produce selective and long-lasting vasoconstriction in the canine external carotid bed (48), but the pharmacological profile of this effect is complex (49). Using GR127935, yohimbine and prazosin as antagonists, we showed that it is mainly mediated by 5-HT1B/1D receptors and α2-adrenoceptors (41); the latter is consistent with subsequent findings showing that both α1- and α2-adrenoceptors mediate the canine external carotid vasoconstriction to adrenaline and noradrenaline (50). Since α2-adrenoceptors include at least three receptor subtypes (α2A, α2B and α2C) (51), we investigated the pharmacological profile of the above responses to ergotamine and dihydroergotamine. Thus, using selective antagonists, such as SB224289 (5-HT1B), BRL15572 (5-HT1D), rauwolscine (α2-adrenoceptors), BRL44408 (α2A), imiloxan (α2B) and MK912 (α2C), it was revealed that the canine external carotid vasoconstriction to dihydroergotamine (52) and ergotamine (53) is mediated by 5-HT1B receptors and α2A/2C-adrenoceptors. Therefore, the canine external carotid bed is an experimental model highly useful for the screening of new compounds with potential antimigraine activity via a vasoconstrictor mechanism, also involving subtype-selective α-adrenoceptor agonists (54).
In vivo human models (i.v. infusion of vasodilator agents)
Several studies have reported that there is an increase in plasma CGRP concentrations associated with cranial vasodilation during a migraine attack (see 18, 55, 56). Thus, the human in vivo models for migraine are based on the intensity and duration of the headache induced by infusions of headache-inducing substances: (i) nitroglycerin, (ii) histamine, (iii) prostaglandin E1, (iv) m-chlorophenylpiperenzine, or (v) CGRP (57). These substances are infused in both migraineurs and non-migraineurs (healthy controls) and the responses are evaluated on the basis of a verbal scoring method (pain intensity) established by the International Headache Society (IHS).
Amongst the substances that induce headache, nitroglycerin is most useful because it affects vascular tone, inflammation, central desensitization and pain transmission (58). The major advantages of using nitroglycerin are: (i) good tolerance, (ii) short half-life, (iii) blood–brain barrier penetration, (iv) ease of administration, and (v) relatively few side-effects. Nitroglycerin produces an immediate, short-lasting and pulsatile headache in healthy volunteers. In contrast, in patients with a history of migraine, a mild to moderate headache is initially observed; nevertheless, a genuine migraine-like headache is observed 5–6 h after nitroglycerin infusion (57). The mechanism of action of nitroglycerin to produce headache involves the release of nitric oxide (NO), a potent vasodilator that plays a role in nociception. The importance of NO as a potential initiator of the migraine attack opens a new avenue for the treatment of migraine and other vascular headaches (57).
Migraineurs seem to be supersensitive to NO donors (57) and the vascular effects of CGRP released from trigeminal nerves, at least in a rat migraine model, are partly mediated by the endothelial release of NO (59). Indeed, i.v. infusions of hα-CGRP in migraineurs can produce a migraine-like headache (58); hence, the trigeminal release of CGRP is a reliable marker for migraine (56). Similar to nitroglycerin, i.v. infusions of histamine in migraineurs produced both an immediate and a delayed headache, which was blocked by the histamine H1 receptor antagonist mepyramine (60). Moreover, the phosphodiesterase-5 inhibitor dipyridamole also produced a mild to severe headache in healthy volunteers (61).
Another model in humans determines the effects of vasodilator substances on the forearm blood vessels (62). Infusions of CGRP in the brachial artery result in a marked vasodilation of the forearm vasculature, as assessed by venous occlusion plethysmography (Fig. 4, left panel) and this vasodilation is, at least in part, dependent on the local release of NO (62). In fact, the vascular beds of the forearm and fingers can be used to study the mechanisms of the antimigraine drugs in the peripheral vasculature (63, 64); in this respect, sumatriptan (Fig. 4, right panel) and some second-generation triptans constrict brachial blood vessels, suggesting that vasoconstriction is an important mechanism behind the therapeutic efficacy of triptans (63, 64). Van Es et al. (63) demonstrated that sumatriptan also reduces arteriovenous anastomotic blood flow in human forearm.
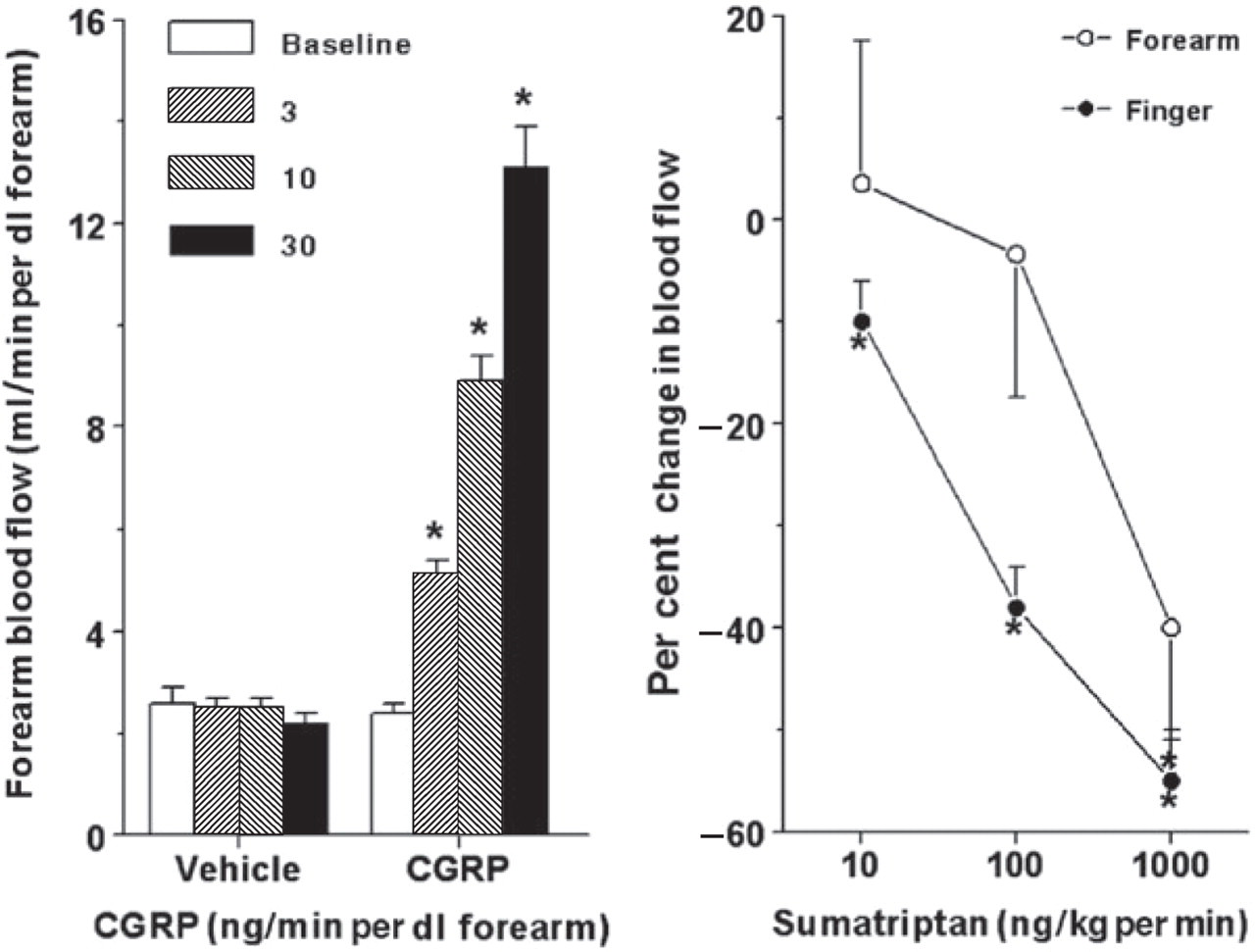
Left panel: Changes in human forearm and finger (right panel only) blood flow to infusions of calcitonin gene-related peptide (CGRP) and sumatriptan, as assessed by venous occlusion plethysmography. ∗P < 0.05 vs. respective baseline values. Data, presented as mean ± SEM, are from De Hoon et al. (62) (left panel) and Van Es et al. (63) (right panel).
Despite the availability of volunteers (healthy as well as migraineurs) and the numerous advantages implicit in human models of migraine, such benefits are limited by practical and ethical issues. Therefore, human isolated blood vessels are increasingly being employed as experimental models.
In vitro human models
These models offer several advantages: (i) drug–receptor interactions at equilibrium; (ii) the possibility of carrying out a detailed pharmacological analysis, mounting multiple segments of blood vessel in parallel; (iii) no influence by pharmacokinetic factors; and (iv) exclusion of central and autonomic mechanisms as well as the effects produced by circulating hormones, distending pressure, etc. Notwithstanding the benefits provided by the in vitro models, these are only complementary to the information obtained with in vivo models (see above). The in vitro models that stand out in characterizing potential antimigraine drugs are isolated cranial (meningeal, temporal, basilar) and coronary arteries; the results obtained from these blood vessels allow one to assess cranial selectivity.
Isolated cranial blood vessels. As previously pointed out, the therapeutic efficacy of acutely acting antimigraine drugs is most probably mainly mediated by constriction of dilated cranial arteries (6, 11, 18). Therefore, this model considers the use of three cranial arteries, namely the middle meningeal, the basilar and the temporal (65–68). The human middle meningeal artery is heavily innervated with afferent sensory fibres containing substance P, neurokinin A and CGRP and originating from the trigeminal ganglion (69). Cumulative concentration–response curves are used to determine the vasoconstrictor potency and efficacy of the prospective antimigraine agent. The rank order of potency of 5-HT receptor agonists in this preparation positively correlates with affinity measurements in cell lines expressing the 5-HT1B receptor (65). Molecular studies have detected mRNA encoding predominantly 5-HT1B receptors, but also other 5-HT receptors such as 5-HT1F, 5-HT2A, 5-HT2B, 5-HT4 and 5-HT7 (70, 71). In line with the above findings, it has been demonstrated that during a migraine attack blood flow velocity in the middle cerebral artery significantly decreases, and this is normalized after administration of sumatriptan (72). Indeed, sumatriptan also constricts cranial vessels in vitro (66, 67, 73). Therefore, studying human isolated cranial blood vessels gives reliable information regarding the behaviour of these vessels in migraine headache and the effects of novel therapeutic agents.
CGRP has also been widely studied by using in vitro human models. In the development of CGRP receptor antagonists, such as BIBN4096BS, as a viable option for acute antimigraine therapy, not only studies in human arteries (74, 75) but also in other species (76–78) help in understanding the mechanism of action of CGRP antagonists. Denudation of the arteries provides information whether the receptors are present on the endothelium or in the vascular smooth muscle of arteries. The in vitro models also provide insight into downstream signalling, as CGRP activates adenylyl cyclase to increase cyclic adenosine monophosphate (cAMP), hence causing vasodilation. This increase in cAMP induced by CGRP is attenuated by the CGRP receptor antagonist BIBN4096BS (75, 79). Further, to investigate the role of endogenous neuropeptides in perivascular nerve endings, artery segments can be stimulated chemically (80) or electrically to release CGRP and other neuropeptides.
Isolated coronary blood vessels. Human isolated coronary arteries are useful in analysing a major potential side-effect of antimigraine drugs, namely, chest symptoms (chest pressure, tightness and pain). Admittedly, the chest symptoms may not be in all cases due to coronary vasoconstriction, but they occur in up to 15% of patients treated with sumatriptan (81, 82). The right epicardial coronary artery is the most commonly used segment for in vitro studies.
It is now well known that triptans constrict coronary arteries, but the effect on cranial vessels, where, contrary to peripheral arteries, the 5-HT1B rather than 5-HT2 receptor dominates, is more marked (see 46, 83). Eletriptan (Fig. 5) (68, 84), sumatriptan (68, 84), rizatriptan (70), frovatriptan (85), almotriptan (86) as well as donitriptan (87) have been demonstrated to be several-fold more potent in contracting the human middle meningeal artery than the coronary artery. More importantly, the magnitude of contraction elicited by triptans in cranial vessels is much more than that in the coronary arteries. Therefore, at therapeutic plasma concentrations, triptans contract cranial vessels much more than the coronary artery (Fig. 5) (68, 84–87). The reason for this is not clear, but it may be related to the higher density of 5-HT1B receptors in the cranial compared with coronary arteries (70).

Concentration response curves of eletriptan on human middle meningeal (•) and coronary (▪) arteries. Superimposed on these curves are the free C max (protein-unbound fraction of the maximum plasma concentration) of eletriptan observed in human subjects after oral ingestion of a single 40-mg or 80-mg tablet. It may be noted that the drug elicits a substantial contraction of the middle meningeal artery, while there is only a minimal effect on the coronary artery (84).
Other studies have analysed the effects of CGRP receptor ligands in the coronary arteries (75, 79, 88). Accordingly, BIBN4096BS, the only CGRP receptor antagonist evaluated in clinical trials for acute antimigraine therapy, does not constrict coronary arteries (75); thus, BIBN4096BS seems to possess a clear advantage over the triptans. However, it is yet to be ascertained whether the antagonism of coronary artery dilation by BIBN4096BS has consequences during pathophysiological conditions, where endogenous CGRP may be important, e.g. cardiac preconditioning (89).
Migraine models based on neurogenic involvement
The basic perception behind the development of neurogenic models for migraine is that migraine pain is due to a sterile neurogenic inflammation within the meninges followed by activation of trigeminal nerve terminals (90) to release neuropeptides (substance P, neurokinin A and CGRP) responsible for some features of migraine (2). Thus, the efficacy of acutely acting antimigraine drugs is believed to be due to a presynaptic action on sensory nerves, thereby inhibiting neuropeptide release (2). Moreover, mechanisms that do not seem to be mediated solely by the 5-HT1B/1D receptor have also been implicated in migraine relief (91). These include inhibition of the trigeminovascular system peripherally and/or centrally (2, 27).
Inhibition of plasma protein extravasation after trigeminal stimulation
The concept of plasma protein extravasation in migraine pathogenesis is based on the observation that plasma protein extravasation is produced by antidromic stimulation of trigeminal ganglia and sensory nerves in rats and guinea pigs (92). Clinically effective antimigraine agents, such as the ergots, triptans, opioids and valproate, inhibited this sterile neurogenic inflammation, suggesting that inhibition of plasma protein extravasation could be predictive of antimigraine therapeutic activity (92). Indeed, sumatriptan was shown to inhibit plasma protein extravasation and this effect was attenuated by the 5-HT1B/1D receptor antagonist GR127935 in both rats and guinea pigs, showing the involvement of 5-HT1B/1D receptor subtypes (90). However, in mice this effect proved to be mediated by the 5-HT1B receptor, whereas in guinea pigs and rats it was a 5-HT1D receptor-mediated response (90, 93, 94). Although triptans have high affinity for both 5-HT1B and 5-HT1D receptor subtypes, they may also act on other subtypes (90). In this respect, plasma protein extravasation was also inhibited by 5-carboxamidotryptamine and CP122288 in 5-HT1B receptor knockout mice and this effect was not prevented by GR127935 in guinea pigs (93, 95). Likewise, CP122288 (which displays high affinity for 5-HT1F receptors) showed a much higher potency than sumatriptan in rats and this did not correlate with its affinity for 5-HT1B or 5-HT1D receptors (93, 95); this suggests that at least part of the CP122288 action may be via the 5-HT1F receptor. Moreover, a number of 5-HT1 receptor agonists that inhibit plasma protein extravasation in guinea pig dura mater display a rank order of potency that correlated with their affinity towards the 5-HT1F rather than the 5-HT1B or 5-HT1D receptor subtype (90, 96). Accordingly, a selective 5-HT1F receptor agonist, LY344864, was developed (96, 97). This compound inhibited plasma protein extravasation following trigeminal ganglion stimulation in rats and was found modestly effective in acute migraine treatment (98). However, it remains unclear whether the two properties are related. LY344864 also inhibited the activation of brainstem neurons in response to the stimulation of dura mater as well as c-fos expression in trigeminal nucleus caudalis (90); this suggests that the primary mechanism of LY344864 is central (i.e. interruption of the ascending pain pathways) rather than peripheral (inhibition of plasma protein extravasation) (90). Interestingly, the selective 5-HT1D receptor agonist PNU-142633F, which blocked plasma protein extravasation in guinea pigs (99), was ineffective in migraine treatment (100). Other studies have shown that plasma protein extravasation can be inhibited by the CGRP receptor antagonist CGRP8-37 (101, 102).
Plasma protein extravasation models do not always predict antimigraine efficacy (2), as clearly evidenced by the failure of several compounds in clinical trials, including: (i) the NK1 receptor antagonist, lanepitant (103); (ii) specific plasma protein extravasation inhibitors, such as CP122 288 and 4991W93 (104); (iii) the ETA/B receptor antagonist bosentan (105); and (iv) the neurosteroid ganaxolone (106). In addition, the clinical antimigraine predictability of plasma protein extravasation assays became questionable following an elegant clinical study in migraine showing no increases in retinal or choroid permeability (107); this contrasts with the increase in retinal or choroid permeability following trigeminal ganglion stimulation in rats (90).
Central trigeminal neuronal inhibition
The importance of the brainstem in migraine pathogenesis is emphasized by its activation during migraine attacks, where blood flow increases in the cerebral hemispheres (cingulate, auditory and visual cortex) as well as brainstem (108). Sumatriptan relieved the headache and reversed the increase in cerebral blood flow, but not in the brainstem, indicating that persistent brainstem activation is due to other factors, including increased activity of the endogenous antinociceptive system. Moreover, brainstem activation may be inherent in the migraine process itself, and continuous activation of the brainstem (despite symptom resolution by sumatriptan) may account for the headache recurrence (108).
Based on this finding, animal migraine models have been developed to study c-fos activation of the trigeminal nucleus caudalis; interestingly, this effect was not altered by sumatriptan (109–111). However, the second-generation triptans, such as zolmitriptan (112), naratriptan (113) and eletriptan (113, 114) as well as dihydroergotamine (115) inhibited the action potentials generated in the trigeminal nucleus caudalis after superior sagittal sinus stimulation in cats and dural stimulation in rats (116). This discrepancy could be due to poor central penetration by sumatriptan (83) compared with second-generation triptans with central inhibitory effects (2). Consequently, it has been argued that the blood–brain barrier may be disrupted during migraine (117); indeed, under experimental disruption of the blood–brain barrier by hyperosmolar mannitol, sumatriptan produced central inhibitory effects (118). However, there is little or no evidence for a disrupted blood–brain barrier based on computed tomography or MRI findings in migraine patients (119, 120).
Several lines of pharmacological evidence indicate that potent antimigraine agents act on the second order trigeminal neurons to reduce cell activity, suggesting that trigeminocervical complex neurons in the caudal brainstem could be a possible target for antimigraine activity (27, 108). It is likely that this central inhibitory effect is mediated by 5-HT1B/1D receptors since the central inhibitory effect of eletriptan in cats is amenable to blockade by GR127935. In addition, the involvement of 5-HT1D rather than 5-HT1B receptors is crucial for this effect (113, 114). Moreover, CGRP mediates sensory nerve transmission between the first and second order afferent input from the cranial blood vessels, and centrally penetrating CGRP receptor antagonists may attenuate sensory nerve transmission (2, 5). Recently, adenosine A1 receptors were localized in human trigeminal ganglia, suggesting a potential usage of adenosine A1 receptor agonists to inhibit trigeminal nociception (13).
A strong link between migraine and the glutamatergic system is suggested on the basis that migraine pain-relay centres contain glutamate-positive neurons and glutamate is implicated in cortical spreading depression, trigeminovascular activation and central sensitization (121). Thus, glutamate receptor-subtype antagonists may be useful in migraine; indeed, some compounds are effective in preclinical models of migraine and probably in the clinic (122, 123). Moreover, the selective NMDA receptor antagonists memantine and MK-801 dose-dependently and significantly reduced c-fos expression induced by capsaicin (124).
The selective blockade of NK1 receptors decreased c-fos response in the trigeminal nucleus caudalis after electrical (125) or chemical (126) stimulation of the trigeminovascular system. Another major molecular marker extensively studied in relation to migraine is the nuclear factor-κB (NF-κB) (127). This factor plays a pivotal role in inducible nitric oxide synthase induction and controls the transcription of the acute-phase proteins (128), which, under basal conditions, sequesters NF-κB within the cytoplasm. In a rat migraine model, infusion of the NO donor glyceryl trinitrate produced activation of the NF-κB in the dura mater, which was suppressed by the anti-inflammatory parthenolide (127). This model based on probing the neuronal effects of NO has provided interesting insights into the neuroanatomical circuits and neuropharmacological mechanisms involved in the initiation and repetition of migraine attacks (7). This is in accordance with the efficacy of anti-inflammatory agents such as parthenolide and aspirin in reducing the frequency and intensity of migraine attacks (129, 130). These molecular markers in combination with in vitro and in vivo techniques may provide crucial insight into migraine pathophysiology.
Central pain sensitization
Burstein and coworkers, who reported that application of an ‘inflammatory soup’ containing histamine, 5-HT, bradykinin an prostaglandin E2 on rat dura activated trigeminal neurons and enhanced their sensitivity to mechanical stimuli, proposed that this chemosensitivity and sensitization is characteristic of some types of headaches and may contribute to throbbing of migraine pain (131). Early sumatriptan intervention (i.e. together with application of inflammatory soup) effectively blocked the development of all aspects of central sensitization (expansion of dural receptive fields, reduction of neuronal response threshold, spontaneous firing rate, increased neuronal response magnitude to skin brushing, and reduced response threshold to skin heating), but late sumatriptan intervention (2 h after inflammatory soup) counteracted only the first two aspects of central sensitization (132). When both peripheral (trigeminal ganglion) and central (medullary dorsal horn) trigeminovascular neuronal potentials were simultaneously recorded in rats, sumatriptan prevented the induction of sensitization and normalized the heightened intracranial mechanical sensitivity following sensitization in central but not in peripheral neurons; however, the drug failed to attenuate the increased spontaneous activity established during sensitization in both neurons (27). These authors concluded that sumatriptan inhibits neither peripheral nor central trigeminovascular neurons directly, but exerts its action via presynaptic 5-HT1B/1D receptors in the dorsal horn to block synaptic transmission between axon terminals of the peripheral trigeminovascular neurons and cell bodies of their central counterparts, thus suggesting that the analgesic action of triptans manifests in the absence, but not in the presence of central sensitization (27).
In agreement with their studies in rats, Burstein and coworkers reported that over 75% of migraine patients develop cutaneous allodynia during migraine within the referred pain areas, initially on the ipsilateral side of the head and later on the contralateral side and on ipsilateral forearm and they hypothesized that the cutaneous allodynia can be used to predict the effectiveness of triptans (10, 133). Indeed, only 15% of patients with, compared with 93% without cutaneous allodynia were rendered pain-free by sumatriptan within 2 h of treatment and similar responses were observed in the two groups whether sumatriptan was administered early or late in migraine attack (132). Interestingly, sumatriptan effectively terminated the throbbing of migraine pain (peripheral sensitization) in the vast majority of patients, even when pain relief was incomplete or allodynia was not suppressed (132).
The value of this model, as indeed of other neurogenic models, is still unclear. So far, only sumatriptan has been studied and one is uncertain if this triptan indeed passes the blood–brain barrier effectively (see 6). However, the stock of this model will increase if a new compound, selected on the basis of this model and devoid of vasoconstrictor properties, is found clinically effective.
Trigeminal primary afferents in vitro Slices of rat trigeminal nucleus caudalis were used to study the release of neurotransmitters, namely CGRP (134). Similarly, slices of rat neocortex were used to study cortical spreading depression induced by exposure to artificial cerebrospinal fluid with elevated potassium chloride (135). In line with this model, ifenprodil, an NR2B receptor subunit-selective NMDA receptor antagonist, suppressed the spreading depression induced in slices of mouse entorhinal cortex (136).
Integrated migraine models
The pathogenesis of migraine appears to be an integrated process involving the trigeminovascular system (11, 18). Accordingly, innovative models of migraine have been developed in which the trigeminal ganglia/nerves are stimulated and the neuroinflammatory processes, such as vasodilation, trigeminal nociception and trigeminal CGRP release, are determined (11, 18). CGRP not only dilates cranial blood vessels, but also transmits vascular nociception (5). The obvious advantage of these models is that they allow the study of both presynaptic and postsynaptic actions of any potential antimigraine compound.
Electrical stimulation of the trigeminal ganglia/nerves and superior sagittal sinus
Electrical stimulation of the trigeminal nerve in humans evokes the release of CGRP in cranial venous blood (2). Moreover, during the headache phase of migraine, the plasma concentration of CGRP, but not of substance P, increases in the jugular venous blood (2). Therefore, CGRP released from trigeminal sensory nerves innervating cranial blood vessels produces vasodilation, thereby causing headache (90, 137, 138). On this basis, several animal models have been developed to demonstrate: (i) cranial vasodilation associated with the trigeminal release of CGRP, and (ii) the inhibitory effects of antimigraine drugs on this parameter. Triptans attenuate cranial vasodilation induced by trigeminal stimulation as well as CGRP release in rats. However, carotid vasodilation in guinea pigs following trigeminal ganglion stimulation is mediated by vasoactive intestinal peptide, which was not amenable to blockade by antagonists at CGRP or tachykinin receptors (139, 140). Therefore, another model was developed in which trigeminal sensory Aδ-fibres that only release CGRP are electrically stimulated and the dural blood vessel diameter is measured by an intravital microscope through a closed cranial window (Fig. 6) (90, 137, 138). Electrical stimulation of this cranial window as well as intravenous infusion of substance P and α-CGRP in rats increase dural blood vessel diameter (90, 94). Interestingly, the NK1 receptor antagonist, RP 67580, clearly antagonized substance P-induced vasodilation, but not the neurogenic vasodilation (90). In contrast, the CGRP receptor antagonist, CGRP8-37, abolished the vasodilation induced by both α-CGRP and neurogenic stimulation (138, 141) (Fig. 6, upper left panel), demonstrating that the neurogenic vasodilation is mediated by endogenous CGRP released from trigeminal sensory nerves. This observation is consistent with clinical data showing that the levels of CGRP, but not those of substance P, are elevated during the headache phase of migraine (2). A recent study on this model showed that BIBN4096BS produced a significant inhibition of both CGRP- and electrically induced increases in the diameter of meningeal arteries, but not of pial and cerebral arteries (142). Significantly, sumatriptan attenuated dural vasodilation following trigeminal stimulation (Fig. 6, right panels), but not the response to CGRP (Fig. 6, lower left panel) (137), probably via presynaptic inhibition of CGRP release. The use of selective antagonists suggests that the above inhibitory effect of sumatriptan is mediated via prejunctional 5-HT1B receptors in rats and 5-HT1D receptors in guinea pigs, cats and humans (90).

Increase in the dural vessel diameter (%) produced by calcitonin gene-related peptide (CGRP) (left panels) and electrical stimulation of the surface of a cranial window (right panels) in the anaesthetized rat before (control) and after CGRP8-37 or sumatriptan. ∗P < 0.05 compared with the control values. Data, presented as mean ± SEM, are from Williamson et al. (137, 138).
Chemical stimulation of sensory nerve fibres with capsaicin
Stimulation of sensory nerve fibres results in the release of neuropeptides in both the central and peripheral nervous system (143). In the former, these neuropeptides transmit nociceptive signals to the spinal cord and brain, whilst in the latter they promote neurogenic inflammation, such as plasma protein extravasation, neuropeptide release and vasodilation. This model allows us to study the inhibitory interactions between antimigraine agents and peripheral trigeminal fibres.
Several studies have shown that sensory nerves innervating the cerebral vasculature contain substance P and CGRP (5); however, capsaicin-induced relaxation of guinea pig isolated basilar artery is mainly mediated by CGRP (144, 145). In this context, in an established porcine migraine model, capsaicin was infused into the carotid artery and the carotid haemodynamic responses as well as trigeminal CGRP release were investigated using BIBN4096BS (146). The results clearly show that capsaicin-induced carotid and arteriovenous anastomotic vasodilation are mediated mainly by CGRP (Fig. 7). Admittedly, as reported earlier (147), vasodilator responses to capsaicin tend to wear off following subsequent infusions of capsaicin, suggestive of tachyphylaxis, which was rather limited, possibly due to a neuronal reuptake of released CGRP into capsaicin-sensitive perivascular nerves (148).

Total carotid conductance (TCC), arteriovenous anastomotic conductance (AVAC) and jugular venous plasma calcitonin gene-related peptide (CGRP) concentrations measured at baseline and following infusions of capsaicin (10 μg/kg per min, intracarotid artery infusion) given in anaesthetized pigs before (control) and after i.v. administrations of BIBN4096BS (100, 300 and 1000 μg/kg). ∗P < 0.05 vs. baseline values; †P < 0.05 vs. response after the corresponding volume of vehicle (data not shown). All data, presented as mean ± SEM, are from Kapoor et al. (146).
Future directions and perspectives
Overall, migraine models have provided significant insights into pathophysiological mechanisms. Indeed, a new era in migraine research has dawned with the revelation of several genetic mutations linked to this neurovascular disorder. Mutations in the P/Q-type calcium channel CACNA1A, Na+/K+ pump ATP1A2 or neuronal voltage-gated Nav1.1 sodium channel SCN1A genes (149–153) can result in familial hemiplegic migraine. A knock-in mouse carrying the human pure FHM-1 R192Q mutation gene has reduced threshold and increased velocity of cortical spreading depression (154). Although the role of these genes is yet to be clarified, incorporating knowledge of the heritability of migraine in existing migraine models may shed further light on the pathogenesis of migraine. Also, a major part of migraine research should be focused on preventive medications, which are believed to affect abnormal neuronal changes in the brain (155). In this respect, putative prophylactic antimigraine drugs, such as botulinum toxin type A (156) and flunarizine (157), may decrease CGRP release from trigeminal neurons and reduce spontaneous synaptic currents in rat neocortex, respectively. These findings suggest that migraine models can be used to study the mechanisms involved in migraine prophylaxis. However, the challenge of finding novel and more effective therapeutic strategies that prevent or control migraine attacks remains open.
The continuing evolution of appropriate migraine models is instrumental in understanding the complex pathophysiology of this syndrome and development of more selective antimigraine drugs. Evidently, this requires an integrative approach that involves the use of several models (rather than only one) so that the advantages add up to the progress and the limitations are minimized. In this respect, the introduction of transgenic animal models and pharmacogenomics will further advance our understanding at genetic and molecular levels. This will ultimately result in new therapeutic options for patients whose migraine attacks at present remain inadequately controlled.