
Abstract
Although not without controversy, an influence of the autonomic nervous system in headache is a matter for current debate. A possible contact site of autonomic and sensory nerves is the dura mater, where they form a dense network accompanying blood vessels. We investigated interactions between autonomic and nociceptive fibres by measuring release of calcitonin gene-related peptide (CGRP) and prostaglandin E2 (PGE2) from the dura mater, in vitro. The parasympathomimetic agent carbachol did not change basal release of CGRP or PGE2, whereas it diminished release induced by a mixture of inflammatory mediators. Norepinephrine did not change induced release of CGRP or PGE2, nor basal release of CGRP. However, basal release of PGE2 was enhanced by norepinephrine, and this enhancement was reduced by serotonin through 5-HT1D receptors. We conclude that sympathetic transmitters may control nociceptor sensitivity via increased basal PGE2 levels, a possible mechanism to facilitate headache generation. Parasympathetic transmitters may reduce enhanced nociceptor activity.
Introduction
The large arteries at the base of the brain and the meningeal (dural) arteries and veins are wrapped in a dense network of nerve fibres. These perivascular fibres belong to three different neuronal systems: the sympathetic and parasympathetic nervous systems, both of which are efferent, and the afferent trigeminovascular system. The various fibres are so close to each other that stimulation of one of the systems may influence the other two systems by neurotransmitter or peptide release (1–3).
The trigeminovascular system of the dura mater has been implicated in headache. The pulsating character of headache has been interpreted as a response of sensitized perivascular nerve fibres to increases in systolic blood pressure (4). Furthermore, neurogenic inflammation of the dura mater has been thought to contribute to the pathophysiology of migraine headache because calcitonin gene-related peptide (CGRP) was shown to be released during migraine attacks, in addition to an increase in regional cerebral blood flow (for review see (5)). Thus, in addition to putative brainstem mechanisms (6), the trigeminovascular system is thought to play a role in the pathophysiology of headache.
In migraine patients dysfunction of the autonomic nervous system has often been found but has remained controversial (see (7–9)). The effectiveness of β-adrenoceptor antagonists (10) and α2 agonists (11, 12) as prophylactic drugs supports a role of the sympathetic nervous system in migraine. Most studies have examined systemic parameters of sympathetic or parasympathetic activity, e.g. autonomic reflexes, blood levels of transmitters, etc. and have compared healthy subjects with migraineurs in various stages of the disease. However, the meningeal vessels might be relatively independent of a general autonomic dysfunction and might rather depend on local processes in the tissue (13, 14). Due to their innervation (see above) the meningeal vessels could be one site of interaction of the nociceptive system and the mediators of the autonomic nervous system. To explore this possibility, we used an established in vitro model (15, 16) to study local processes in the dura mater. We assessed whether the release of CGRP and prostaglandin E2 (PGE2) in the dura can be influenced by mediators of the sympathetic and parasympathetic nervous system.
CGRP is of interest because it is released from nociceptive primary afferent neurons following depolarization (thus it is an indicator of nociceptor activation) and because it causes vasodilation during neurogenic inflammation. It may also act directly on CGRP receptors on primary afferent neurons (17). Recently, the non-peptide CGRP antagonist BIBN4096BS has been shown to reduce migraine pain effectively in clinical trials, thus underscoring the role of CGRP in the pathophysiology of migraine (18–20). The inflammatory mediator PGE2 is also released during nociceptor activation, probably from tissue surrounding the afferent fibres (21) and eventually from postganglionic sympathetic terminals (22; not explored for the dura mater). If there is interaction between primary afferents and nearby autonomic efferents, the basal or evoked release of CGRP and PGE2 should be influenced.
Parasympathomimetics such as acetylcholine (ACh) either facilitate or inhibit CGRP release and nociceptor activity (23–28). The effect depends on the acetylcholine concentration and the localization and number of inhibitory muscarinic ACh (mACh) and excitatory nicotinic ACh (nACh) receptors (29, 30). Effects of parasympathomimetic agents on PGE2 release have not been studied. Putative effects of sympathetic mediators on sensory fibres in the trigeminovascular system are not known. In the skin of intact rats norepinephrine (NE) did not activate nociceptors but after chronic inflammation C-fibres showed sensitivity to NE (31). The action of NE on CGRP and PGE2 release seems to be tissue specific, and inhibitory rather than excitatory effects have been described (32–34).
In the present experiments we tested whether the parasympathomimetic agent carbachol and NE influence basal and stimulated release of CGRP and PGE2 from meningeal tissue in an in vitro model. Because NE is colocalized with serotonin (5-hydroxytryptamine, 5-HT) in sympathetic fibres (for review see (35)) and because of the highly important role of 5-HT1B/D receptors in migraine therapy, the influence of serotonin on the effects of NE on CGRP and PGE2 release was also tested.
Materials and methods
Preparation
Female Wistar rats (n = 82; 243 ± 48 g, raised at the University of Jena) were killed in a CO2 atmosphere. The animals were decapitated and the skin and galea were retracted from the skull. The skull was cut along the saggital suture with a fine saw and divided into halves which were similarly treated. The two hemispheres were removed without injuring the meninges underneath. The skull cavities were then washed for 30 min at room temperature by superfusion with carbogen gased synthetic interstitial fluid (SIF; 108 m
Sampling and stimulation
The skulls were filled with SIF (37°C, pH 7.4). The fluid was removed and pooled from the cavities after 5 min (sample 1). For measuring of CGRP 200 μl of the sample was mixed with 50 μl of enzyme immunoassay (EIA) buffer (see below) and frozen at −20°C. The rest of the sample was immediately frozen at −20°C for PGE2 determination. The whole procedure was repeated for sample 2. The third solution which was applied to the skulls contained the stimulus solution (sample 3). For samples 4 and 5 SIF alone was applied for 5 min each to the cavities.
Experiments were performed to investigate changes either in basal or evoked release following either carbachol or NE application. To evoke release of immunoreactive CGRP (iCGRP) and immunoreactive PGE2 (iPGE2), stimulation with inflammatory mediators (IM, see below) was chosen in order to stimulate sensory afferent fibres without directly activating autonomic fibres. The IM solution was successfully used in a previous study (15).
Solutions
The solutions were: (i) a mixture of IM: bradykinin, histamine, 5-HT in SIF, 10−6
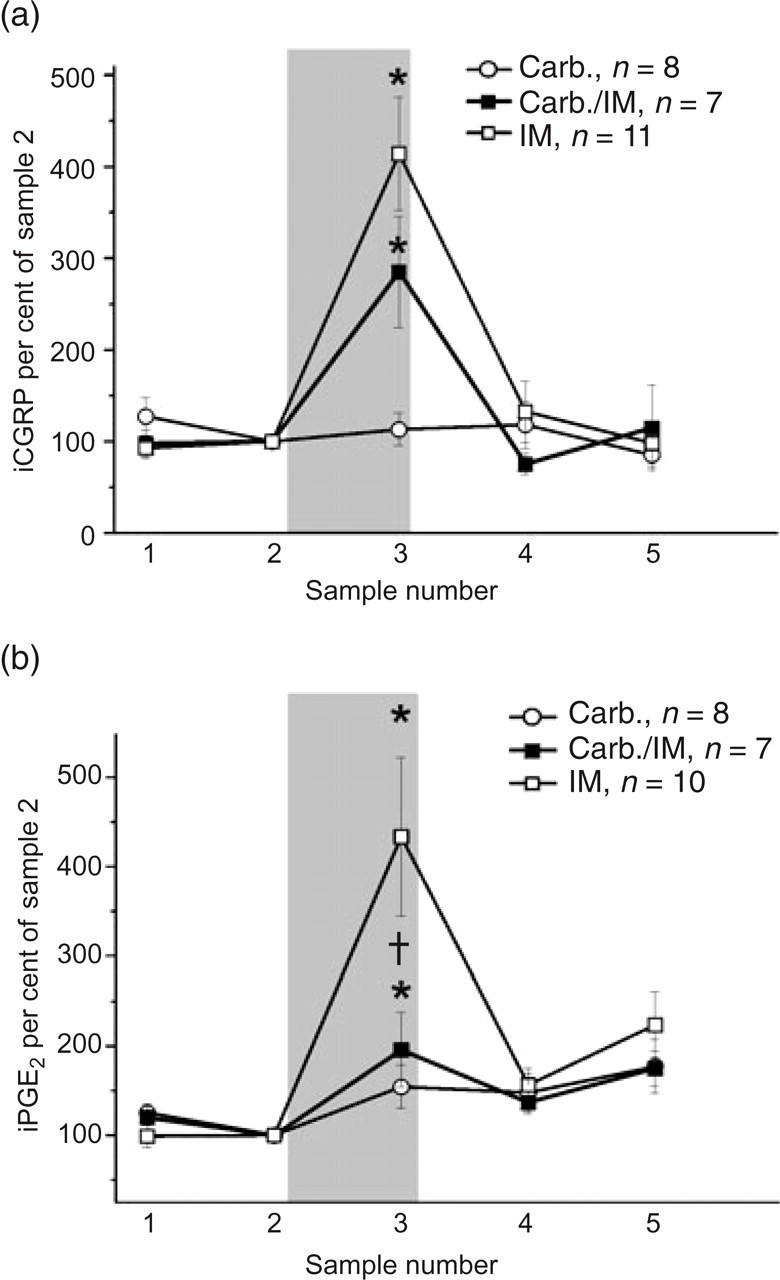
Effect of carbachol (Carb.) on basal and evoked release of immunoreactive calcitonin gene-related peptide (iCGRP) and immunoreactive prostaglandin E2 (iPGE2) from rat dura mater. (a) Carbachol has no effect on basal release of iCGRP, but reduces by 31% the release evoked by inflammatory mediators (IM). (b) Carbachol has no effect on basal release of PGE2 but reduces by 82% the release evoked by IM. Values in the figure are given as percent of the mean concentration in sample 2. The grey area indicates the application of Carb., Carb./IM or IM in sample 3. ∗Statistically significant differences (P < 0.05) between sample 2 and sample 3 in the same preparation, Wilcoxon matched pairs signed rank test, P < 0.05; †statistically significant differences (P < 0.05) between the groups stimulated with IM and Carb./IM in sample 3, Mann–Whitney U-test. n, The number of rats tested.

Effect of norepinephrine (NE) on evoked release of immunoreactive calcitonin gene-related peptide (iCGRP) and immunoreactive prostaglandin E2 (iPGE2) from rat dura mater. Display as in Fig. 1. (a) Application of NE did not change iCGRP release evoked by inflammatory mediators (IM) pH 7.2 or IM pH 5.8. (b) Application of NE did not change iPGE2 release evoked by IM pH 7.2 or IM pH 5.8.

Effect of norepinephrine (NE) and 5-hydroxytryptamine (5-HT) on basal release of immunoreactive calcitonin gene-related peptide (iCGRP) and immunoreactive prostaglandin E2 (iPGE2) from rat dura mater. Display as in Fig. 1. (a) Basal release of iCGRP was not influenced by application of NE, 5-HT or both to the dura mater. (b) Basal release of iPGE2 was massively enhanced by NE. Application of 5-HT together with NE caused a reduction in this effect. This reduction was attenuated by additional application of the 5-HT1D receptor antagonist BRL 15572 (BRL).
Enzyme immunoassay
For the determination of iCGRP an EIA with a minimum detection limit of 2 pg/ml was used (SPIbio Company, Montigny le Bretonneux, France). For the determination of iPGE2 the samples were diluted fivefold with aqua bidest to allow measurements within the highest sensitivity of the assay. The EIA for iPGE2 (Cayman Chemical Co., Ann Arbor, MI, USA) had a detection level of 15 pg/ml. None of the stimulation solutions and drugs interfered with the assay.
Data analysis
The amounts of CGRP and PGE2 released were calculated as pg/ml eluate. The weight of the tissue could not be determined because of the tight adherence of the dura mater to the bone. The data were normalized by defining as baseline the concentration before stimulation (concentration of sample 2, 100%). Results from groups of identical experiments are given as mean ± SEM. Differences within one group of experiments were analysed using the Wilcoxon matched pairs signed rank test. The Mann–Whitney U-test was used for comparisons between two groups. Significance level was P < 0.05.
Results
The basal release (sample 2) from the rat dura mater was 51.28 ± 6.6 pg/ml for iCGRP (n = 82) and 689.8 ± 88.9 pg/ml for iPGE2 (n = 68). In contrast, no iCGRP or iPGE2 was detected in synthetic interstitial fluid alone (negative control, detection level for iCGRP < 2 pg/ml and for iPGE2 < 15 pg/ml). Stimulation with IM, pH 5.8 caused a significant increase of iCGRP, reaching a total concentration of 302.2 ± 75.3 pg/ml (n = 11), and of iPGE2, reaching 2691.6 ± 815.1 pg/ml (n = 10) (Fig. 1a,b).
Effect of carbachol on basal and evoked release of iCGRP and iPGE 2
iCGRP
The application of carbachol 10−5
iPGE 2
The effect of carbachol on the release of iPGE2 resembled its effect on iCGRP. While carbachol alone did not change the release of iPGE2 with respect to baseline, it significantly reduced the iPGE2 release stimulated by IM (pH 5.8) by 82% (Fig. 1b).
Effect of NE on the evoked release of iCGRP and iPGE 2
iCGRP
The application of IM at pH 5.8 increased iCGRP release more than fourfold above baseline (P < 0.05). When IM at pH 7.2 was applied, the increase was about twofold. Evoked iCGRP release was not significantly changed when NE 10−5
iPGE 2
Evoked iPGE2 release was not significantly changed when NE 10−5
Effect of NE and 5-HT on the basal release of iCGRP and iPGE 2
iCGRP
Release of iCGRP was not increased above baseline by the application of either NE 10−5
iPGE 2
5-HT did not change the release of iPGE2 with respect to baseline. In contrast, NE applied to the dura mater enhanced iPGE2 release massively, outlasting the application. In sample 3 the iPGE2 concentration reached 2470.4 ± 1031.9 pg/ml (P < 0.05). When a mixture of 5-HT and NE was applied, iPGE2 release was 42.3% less than with NE alone. Concomitant application of the 5-HT1D receptor antagonist BRL 15572 blocked this effect of 5-HT in sample 3. However, in samples 4 and 5 iPGE2 release was still significantly above basal values but much weaker than with NE stimulation alone (Fig. 3b).
Discussion
In an in vitro preparation of the dura mater carbachol showed no influence on the basal release of iCGRP and iPGE2 but reduced stimulated release. NE influenced neither the basal release of iCGRP nor the stimulated release of iCGRP and iPGE2 from the dura mater. However, NE significantly enhanced basal iPGE2 release. This enhancement was reduced to a half by 5-HT, an effect due at least partly to an action at 5-HT1D receptors. These data show that parasympathetic mediators may have an inhibitory influence on dural nociceptors when these are activated. Furthermore, they show that sympathetic mediators may elevate PGE2 release well above basal levels.
Comparison of the negative controls (SIF) with values of the unstimulated tissue showed basal release of iCGRP and iPGE2 from the dura mater. The concentrations found are in agreement with values that other authors have found in the same preparation (37). While CGRP is stored in vesicles in nociceptive C-fibres and released upon stimulation, iPGE2 might not only be of neuronal origin. Denervation experiments show that in rat skin released iPGE2 originates mainly from non-neuronal tissue (21). This is likely to be true also for the dura mater preparation, since the enhanced release of iPGE2 by NE was not associated with an enhanced release of CGRP from primary afferents measured in solutions taken from the same samples.
Carbachol
Carbachol application was chosen to imitate activity of the parasympathetic fibres. Like ACh, this stable ligand binds to nACh receptors as well as to mACh receptors. Dural ACh receptors have not yet been precisely localized. In the trigeminal ganglion immunofluorescence for mACh and also for nACh receptors is seen in about 20% of the small to medium-sized neurons (29, 30, 38) and nicotine activates a subset of trigeminal ganglion neurons (39). In our hands basal release of iCGRP (and iPGE2) was not changed by the application of carbachol to the dura mater. Thus, carbachol did not directly activate sensory fibres in the dura mater. While data from the literature also do not suggest direct stimulation of dural afferents by carbachol, indirect activation of sensory fibres may occur. Dura mater stimulated by carbachol released histamine, probably from stimulated mast cells (40), and infusion of carbachol into the common carotid artery induced plasma protein extravasation in the ipsilateral dura mater (41).
However, in the present preparation we found inhibitory effects of carbachol. When the release of iCGRP had been increased by IM, carbachol had an inhibitory effect upon dural afferents. The IM-evoked release of iPGE2 was also reduced in the presence of carbachol. In fact, its inhibitory effect was much more prominent on evoked iPGE2 release than on iCGRP release (82% vs. 31% reduction), suggesting that non-neuronal mACh receptors might diminish iPGE2 release in addition to the reduced nociceptor activation.
The action of ACh on evoked iCGRP from rat buccal mucosa and skin is known to be concentration-dependent (20, 30). Low concentrations of ACh (<10−4
Norepinephrine
The basal and stimulated release of iCGRP from dura mater were left unchanged by additional NE application, a phenomenon also observed for the basal and electrically evoked release of iCGRP in a rat skin/nerve preparation (33). However, this may be a tissue-specific feature, since adrenoceptor activation reduced evoked CGRP release in isolated perfused lung (32, 42), cardiac tissue (43) and dental pulp (34, 44, 45). Thus, the number and localization of different adrenoceptors seem to be of importance for iCGRP release. In addition, the condition of the tissue may also be relevant. C-fibres of the skin of normal rats do not respond to NE but NE causes an excitation of the neurons in rats with inflammation (31). Comparable effects were also seen in other models (46–49). In the present experiments, care was taken to avoid any inflammation of the dura mater and the basal levels of CGRP were low, indicating non-stimulated conditions. Thus, under normal conditions primary afferent fibres of the dura mater are probably not directly influenced by NE released from the sympathetic nervous system.
However, NE massively enhanced PGE2 release from the unstimulated dura mater, an effect outlasting NE application. This finding suggests that NE may act indirectly on dural afferents. The sustained PGE2 level might induce an inflammation in the dural tissue and in this way, transmitters from the sympathetic nervous system might slowly change the sensitivity of dural afferents (50). A release of PGE2 by NE was not observed in rat skin (33) but was observed in the hypothalamus, where NE indirectly activates PGE2 synthesis by activating nitric oxide production (51). Surprisingly, in the dura mater the release stimulated by inflammatory mediators was not enhanced by additional application of NE. This might possibly depend on the presence of 5-HT in the stimulation solution (see below).
Because NE and 5-HT are located in the same vesicles and are released together upon stimulation of dural sympathetic fibres (35), we tested the effect of 5-HT, and also of 5-HT plus NE, on iCGRP and iPGE2 release. 5-HT alone did not influence the basal release of iCGRP and iPGE2. A similar observation had been made in the same model and in cultured trigeminal neurons (37, 52). However, the mixture of NE and 5-HT elicited only half of the amount of iPGE2 release compared with the release evoked by the application of NE alone. This effect was lost when a 5-HT1D antagonist was added to the mixture, suggesting that the cotransmitter 5-HT is able to control the effect of NE via inhibitory 5-HT1D receptors. Since most iPGE2 is presumably not released from sensory neurons but from other structures (see above), the 5-HT1D receptors involved should not only be found on sensory trigeminal neurons and in the trigeminal tract where they have been described to be colocalized with CGRP (53–55). Indeed, the inhibition of NE release by activation of 5-HT1D receptors in the dura mater shows that this receptor may also be located on dural sympathetic efferents (56). This would imply an (auto)regulation of sympathetic transmitter release. The basal amount of PGE2 in the tissue might therefore depend on the ratio of NE and 5-HT released from sympathetic terminals.
In conclusion, the present data show that the sensitivity of dural afferents can be tuned by the mediators released from autonomic nerve fibres. In particular, tonic and/or stimulated activation of the sympathetic nervous system might condition receptor sensitivity in the dura mater by releasing local PGE2. However, for the final effect of the sympathetic system on PGE2 release, the ratio of released 5-HT and NE seems to be important. By contrast, parasympathetic transmitter release might reduce CGRP and PGE2 release from the dura mater during periods of sensory activation. Thus the dural vessels and their innervation might be one site where the autonomic nervous system interacts with the nociceptive system.
Footnotes
Acknowledgements
We thank Mrs Helga Müller for expert technical assistance. The project was supported by IZKF Jena (B378-10102).