
Abstract
Calcitonin gene-related peptide (CGRP) and related peptides may be involved in migraine pathogenesis. To understand their vasomotor role in the cerebral circulation, we performed two studies, a pressurized arteriography study of the middle cerebral artery (MCA) and a genuine closed cranial window (gCCW) in vivo study. Using the pressurized arteriography model rat MCAs were mounted on micropipettes, pressurized to 85 mmHg and luminally perfused. The diameter responses to luminally and abluminally applied rat-αCGRP, rat-βCGRP, amylin and adrenomedullin were compared with the resting diameter. Only abluminally applied CGRP induced dilation of the cerebral arteries; E max for αCGRP and βCGRP were 35 ± 0.5% and 10.8 ± 0.2%. These responses were blocked by CGRP8-37. The gCCW model allowed videomicroscopic visualization of the pial vessels in anaesthetized rats. Changes in vessel diameter to intravenously administered αCGRP and βCGRP were compared with pre-infusion baseline. Intravenous infusion of αCGRP and βCGRP in the highest dose induced dilation of the cerebral cortical pial arteries/arterioles of 40.3 ± 7.5% and 49.1 ± 8.4%, respectively. However, this was probably secondary to a decrease in blood pressure of 44.8 ± 3.3 mmHg and 49.2 ± 3.3 mmHg. Our results suggest that CGRP receptors are probably functional on the smooth muscle cells and not on the endothelium of rat cerebral arteries.
Keywords
Introduction
Calcitonin gene-related peptide (CGRP) is a 37 amino acid neuropeptide, first identified in 1982 (1). It belongs to a family of peptides, which also includes calcitonin, adrenomedullin and amylin. Localization studies have shown a wide distribution of CGRP immunoreactive structures in the peripheral and in the central nervous system (2, 3). CGRP-containing nerves innervate blood vessels in various regions and in which CGRP is a potent vasodilator (4). Within the cerebral circulation, there is a dense supply of CGRP-containing nerve fibres originating from the trigeminal ganglion (5, 6) and the peptide may be involved in the pathophysiology of primary headaches (7).
CGRP exists in two isoforms, α and β. Calcitonin and αCGRP are transcribed from the same gene, while βCGRP is formed from a duplication of the calcitonin αCGRP gene (8).
Cloning efforts have resulted in the molecular identification of a CGRP receptor, the calcitonin receptor-like receptor (CRLR). CRLR is a Gs-coupled seven-transmembrane domain receptor (GPCR), which shares 55% sequence identity with the calcitonin receptor (9). Subsequently, it was demonstrated that functional CGRP and adrenomedullin receptors are both derived from CRLR and that the phenotype is determined by co-expression with a particular receptor activity modifying protein, RAMP (10). Co-expression of CRLR with RAMP1 results in CGRP receptor pharmacology while RAMP2 co-expression with CRLR produces an adrenomedullin receptor and with RAMP3 a possible combined receptor. Furthermore, the RAMPs enable expression of CRLR on the cell surface (11). CRLR internalization following CGRP stimulation has been shown to occur together with RAMP1, and both proteins are targeted to the protein degradation pathway (12). RAMP proteins have, in addition, been shown to modulate the pharmacology of the calcitonin receptor, which, in combination with RAMP1 or RAMP3, bind amylin with high affinity (13, 14). In addition to the RAMPs, the CGRP receptor may require another accessory protein for functionality, the receptor component protein (RCP). CRLR and RAMP1–3 have been shown to be localized on the smooth muscle cells of the brain vessels (15).
This study was performed to understand the site of action of CGRP in the cerebral vessel wall. We applied two different models: an in vitro pressurized arteriography model investigating the middle cerebral artery (MCA) and an in vivo genuine closed cranial window (gCCW) studying the cortical pial arteries/arterioles. Furthermore, the study was performed to obtain important data on the prospect of future use of CGRP receptor antagonists in the treatment of migraine (16).
Materials and methods
Tissue preparation
Male Sprague-Dawley rats (250–300 g) were anaesthetized using CO2 and decapitated. The brain was immediately removed and placed in cold (4°C) buffer solution of the following composition (m
Pressurized arteriograph
A section of the MCA (1–2 mm in length) was mounted in a pressurized arteriograph (Living System, Burlington, VT, USA) as previously described (17, 18). Micropipettes were inserted into both ends of the MCA and secured in place with 11–0 nylon ties. The MCA was bathed in the described buffer solution (37°C) equilibrated with a gas mixture consisting of 5% CO2−95% O2, resulting in a pH of 7.4. Transmural pressure of the MCA was maintained at 85 mmHg by raising reservoirs connected to the micropipettes to the appropriate height above the MCA. Luminal perfusion was adjusted to 100 µl/min, ranging from 80 to 100 µl/min, by setting the two reservoirs at different heights. Pressure transducers on either side of the MCA provided direct measurement of perfusion pressure across the MCA. The vessel was magnified 600-fold using a microscope coupled to a digital camera (Axis®; Axis Communication AB, Lund, Sweden) connected to a computer. The program Mary® (Forget-IT, Sweden) saved the pictures every second during the experiment as well as measuring the diameter of the vessels.
The preparation consisted of two separate compartments, the luminal perfusate and the abluminal bath. Adding agonists to the luminal perfusate selectively stimulated receptors on endothelial cells (EC); adding the agonists to the abluminal bath mainly stimulated receptors on the vascular smooth muscle cells (VSMC) (17).
Experimental protocols were not initiated until the MCA diameter was stable for a 15-min period. Any MCA that did not develop spontaneous tone of at least 20% (range 25–35%) compared with the initial diameter within 1 h was excluded from the experiment. The presence of a functional endothelium was tested by luminal administration of adenosine triphosphate (ATP) (10−5
To test the vascular response to stimulation of endothelial receptors, CGRP, adrenomedullin or amylin were added to the luminal perfusate in the concentration range 10−12−10−6
To characterize the response to CGRP stimulation, CGRP was used either alone or in the presence of CGRP8−37, 10−6
Cranial window in vivo preparation
All experimental procedures were performed in accordance with guidelines and regulations for animal care and treatment. The study protocol was approved by The Danish Animal Experimentation Inspectorate (file 2001/561–390).
The experiments were conducted in male Sprague-Dawley rats (300–400 g). Animals remained anaesthetized throughout the experiment. Pentobarbital (Mebumal®, 60 mg/kg) was given intraperitonally for induction of anaesthesia and this was maintained by continuous i.v. infusion of pentobarbital (Mebumal®, 20 mg kg−1 min−1). A tracheotomy was performed and the animals were artificially ventilated (Abovent 7025; Ugo Basil, Comerio, Italy) with a 30/70% mixture of O2/N2O. The body temperature of the animal was kept at 37.0–37.5 °C using an automatically regulated heating plate (Letica HB101; Panlab, Barcelona, Spain). The femoral arteries and veins were cannulated bilaterally with polythene catheters for measurement of mean arterial blood pressure (MABP) (Transducer TCM4-7; World Precision Instruments Inc., Sarasota, FL, USA), arterial blood samples, infusion of anaesthetics and test substances. Arterial blood analysis (ABL520; Radiometer A/S, Brønshøj, Denmark) was performed at least three times during each study. PaCO2, PaO2 and pH were kept within normal limits by adjustments of respirator stroke rate, pH 7.40 ± 0.01, PaCO2 38.6 ± 0.5 mmHg and PaO2 120.4 ± 3.0 mmHg. The animal was placed in a stereotactic frame. Skin and connective tissue were removed from the dorsal side of the skull. The right parietal bone was thinned until transparency using a dental drill. During the drilling, cooling was obtained by applying ice-cold isotonic saline.
Video microscopy
The obtained cranial window was covered with mineral oil (37°C). A cortical pial artery/arteriole was visualized using a video-microscope (Sony DSP digital camera, MS50 objective; Japan). The real-time image was displayed on a TV screen. The diameter of the vessels was continuously measured by a video dimension analyser (V94; Living Systems Instrumentation, Burlington, VT, USA) (19). All measurements and data collection were continuously displayed on a computer monitor by data acquisition and analysis software (Perisoft® version 1; Perimed AB, Järfälla, Sweden).
The experimental model was left stabilizing for 1–1.5 h before starting the study protocols. Two dose–effect protocols were performed: one for αCGRP and one for βCGRP, only one semilogarithmic dose–effect curve was obtained in each animal. The infusion of increasing CGRP doses (0.01 ng/kg to 3 µg/kg) was administered as a bolus of 100 µl, 10 min apart. Every bolus was succeeded by a 150-µl isotonic saline flush.
Statistical analysis
Data are expressed as mean ± SEM unless otherwise indicated. For experiments performed on the pressurized arteriograph, changes in measured diameters of the vessel segments are expressed as a percentage of the resting diameter. Statistical analysis was performed using an unpaired t-test. The diameter of the pial arteries in the in vivo model was measured in arbitrary units (AU); the changes were calculated as the peak percentage changes from the preinfusion baseline. MABP was expressed in mmHg. Comparison between the two subtypes of CGRP and the haemorrhage-induced hypotension was performed using an unpaired t-test. The summary measurements used for the t-test were the mean peak per cent increase from prestimulation baseline and AUC calculated as changes in the first minute after application of stimuli. P-values < 0.05 were considered significant.
Drugs
ATP, αCGRP and βCGRP, αCGRP8−37, adrenomedullin and amylin were obtained from Sigma (St Louis, MO, USA) and Neosystem (Strasbourg, France). Stock solutions of the drugs were made following manufacturer's instructions and stored frozen in small aliquots until use. For the in vitro experiments, drugs were diluted in the described buffer solution immediately before use. All chemicals were obtained from Merck (Darmstadt, Germany). Only double distilled water was used throughout the in vitro experiments. In the in vivo experiments the stock solutions were further diluted in isotonic saline (0.9% NaCl) prior to infusion.
Results
Arteriograph experiments
The mean baseline diameter of the examined blood vessels was 109.6 ± 11.5 µm (mean ± SD) after initial pressurization and 73.9 ± 9.4 µm (mean ± SD) after development of spontaneous tone (n = 28, Fig. 1A,B). The spontaneous myogenic tone was 32.1 ± 8.7% of the initial vessel diameter. ATP (10−5

Illustration of the in vitro studies. (a) Baseline tone just after mounting of the vessel. (b) Following perfusion, tone is induced via a shear stream. Tone just before (c) and (d) when adenosine triphosphate (10−5
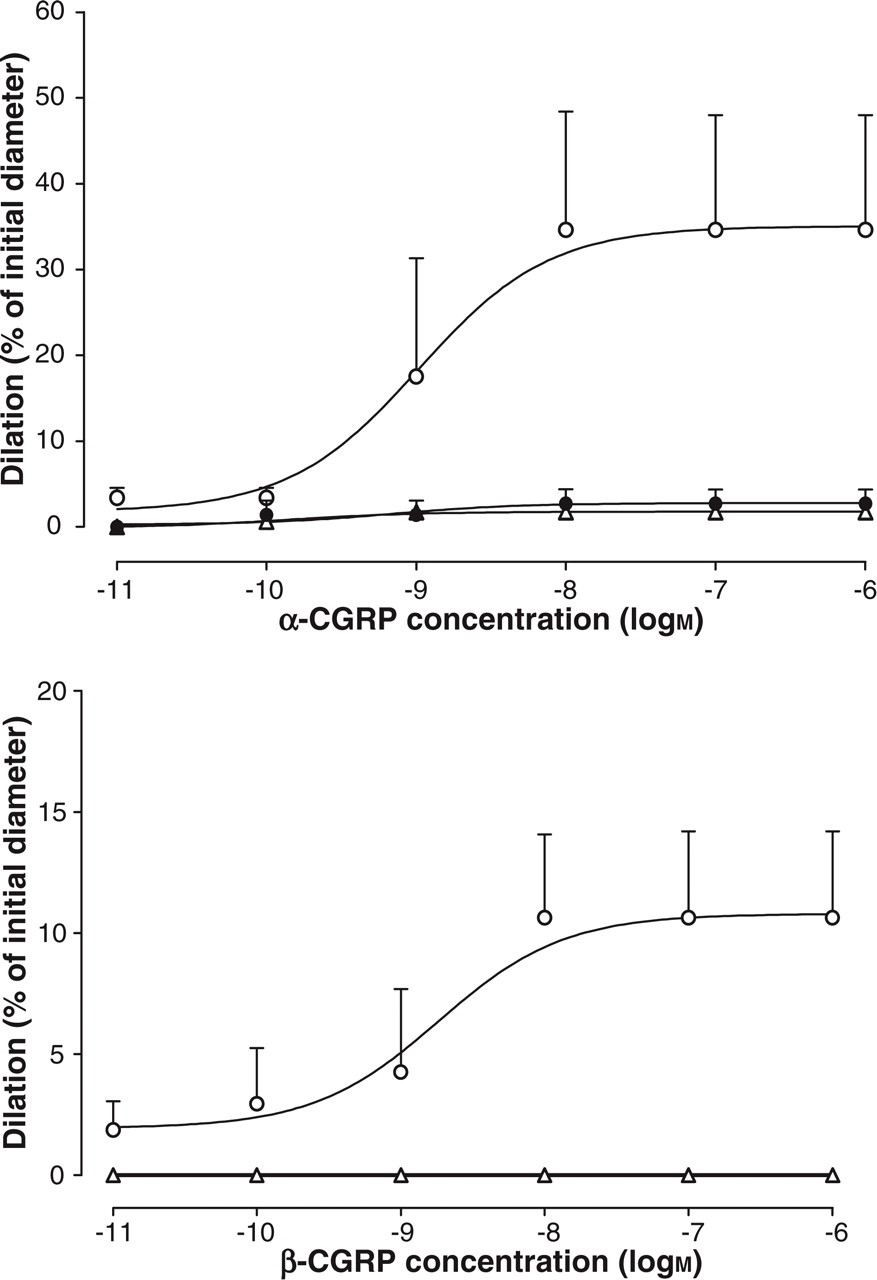
Concentration–dilation curves for luminally (▵) or abluminally (○) administered αCGRP and βCGRP. In the upper graph αCGRP8−37 (d) was found to block the response to αCGRP.
Closed cranial window experiments
Twelve experiments were performed, in which either αCGRP (n = 6) or βCGRP (n = 6) was administered. The mean diameter of the pial vessels was 56.7 ± 5.3 A

Dose–effect curves for αCGRP (▪) and βCGRP (▴). (a) Percentage pial diameter changes. (b) Mean arterial blood pressure (MABP) changes (mmHg).
Increase in pial diameter after CGRP and haemorrhage-induced hypotension is compared in Table 1. Data on haemorrhage-induced hypotension are historical (20). To make a descriptive comparison more obvious the change induced by CGRP was grouped according to the degree of the hypotension, similar to our previous results. A statistical comparison between these data and the present was performed using an unpaired t-test; however, the risk of type II errors should been taken into account. Comparing the effect of haemorrhagic hypotension and systemic αCGRP or βCGRP infusion there was no significant difference in the mean peak response (Fig. 4 and Table 1) or in the calculated AUC.

Histogram displaying percentage changes in pial artery diameter vs. grouped mean arterial blood pressure (MABP) changes. (a) αCGRP and (b) βCGRP. Haemorrhage-induced (▪) and CGRP-induced pial diameter increase (□). ∗∗P = 0.04, comparison between haemorrhage- and CGRP-induced changes (unpaired t-test).
Changes in parameters due to hypotension
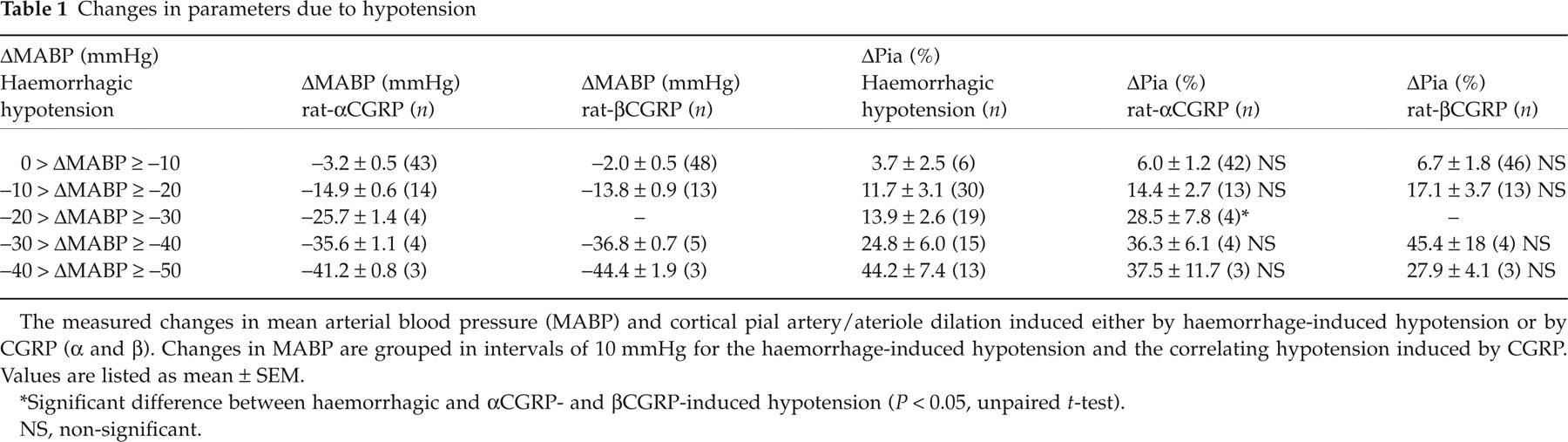
The measured changes in mean arterial blood pressure (MABP) and cortical pial artery/ateriole dilation induced either by haemorrhage-induced hypotension or by CGRP (α and β). Changes in MABP are grouped in intervals of 10 mmHg for the haemorrhage-induced hypotension and the correlating hypotension induced by CGRP. Values are listed as mean ± SEM.
Significant difference between haemorrhagic and αCGRP- and βCGRP-induced hypotension (P < 0.05, unpaired t-test).
NS, non-significant.
Discussion
The present findings on the perfused MCA demonstrate for the first time that CGRP, amylin and adrenomedullin do not cause dilation when applied intraluminally to the isolated rat MCA. A concentration-dependent relaxation was, however, shown when αCGRP or βCGRP were applied abluminally. This response was blocked by CGRP8−37, indicating the presence of a CGRP receptor, which is in agreement with previous studies on isolated ring segments of the cerebral arteries (21). In the in vivo experiments, both subtypes of CGRP induced a dose-dependent increase of the pial diameter after systemic administration. This increase is believed to be mainly a result of regulatory mechanisms activated by the pronounced hypotension seen during the infusion of αCGRP and βCGRP in high doses, since the same effect is seen upon haemorrhage-induced hypotension (20).
During attacks of migraine and cluster headache, an associated peripheral release of CGRP has been observed (22–24). In addition, following sumatriptan administration, the plasma levels of CGRP returned to control levels with successful amelioration of the headache. In migraine-prone individuals, CGRP is able to elicit both an immediate non-migraine headache and, with a delay of 1–12 h, a headache fulfilling the International Headache Society criteria for migraine (25).
In cerebral vasospasm occuring after subarachnoid haemorrhage, the constricted vessel tone could be normalized by systemic administration of αCGRP, while the peptide had no effect on the non-spastic cerebral arteries on the non-affected side (26). These findings indicate a pivotal role of CGRP in limiting the ischaemia induced by the vasospasms. However, at the same time the peptide seems to play a causative role in the initiation of migraine attacks.
The site of receptor interaction is believed to be the VSMCs (27, 28). The sensory perivascular nerves storing CGRP originate in the trigeminal ganglion, as revealed by histochemistry in denervation experiments (6) and by retrograde tracing studies (29). Activation of this system in the cat or humans results in release of CGRP and in an increase in local cortical blood flow (23) that can be attenuated by a CGRP receptor antagonist (30, 31). Our findings with the pressurized arteriograph model are in agreement with an abluminal site of CGRP action and not with an endothelium-mediated relaxant effect. This is supported by studies of isolated cerebral arteries, which relax equally well before and after removal of the endothelium (32). Abluminal application in the arteriograph study further demonstrated that the response to αCGRP was strong and that the CGRP antagonist could block this effect. A similar response was seen after βCGRP, whereas the effects of adrenomedullin and amylin were small.
The pial arteries chosen for the in vivo model were not the MCA, but small arterioles on the cortical surface; however, are most likely branches of the MCA. Hence the correlation between the two models is based on the assumption of a similar anatomical structure and the presence of a blood–brain barrier. Therefore, when administering the peptide intravenously, CGRP would only have access to the luminal side of the arteries. The vasodilatory response obtained was comparable to the E max achieved by abluminal application in the in vitro model. Furthermore, an equal response was achieved for αCGRP and βCGRP, in contrast to the larger effect of αCGRP seen in the isolated MCA.
A previously performed validation study of the gCCW model reported a significant correlation between haemorrhagic induced hypotension and pial arteriolar reactivity (20). When comparing the reactivity of the pial vessels from these historical data with the present, no significant difference was found. This indicates that the pial arteriole diameter increase is not a direct effect of the CGRP, but a secondary haemodynamic regulatory mechanism activated by the induced hypotension.
This leads to a conclusion comparable to that suggested in the perfusion arteriography part of the study: that CGRP-mediated vasodilation is not caused by interaction with a luminally situated receptor but more likely by an abluminal receptor on the smooth muscle cells. An abluminal activation of the CGRP receptor in the closed cranial window model can be achieved by electrical stimulation. Several reports on neurogenically induced dural vasodilation have been published using the described window model (33, 34), but none has so far described pial artery/arteriole responses. In a preliminary study (unpublished data), electrical stimulation of the cranial window caused a dilation of pial arteries. However, future studies using selective CGRP antagonists are necessary to draw final conclusions about the abluminal receptor in the in vivo model.
In summary, we observed that, in vitro, both subtypes of CGRP and to a lesser extent adrenomedullin and amylin interact only with receptors that are present on the smooth muscle cells. These findings were partly supported by experiments with CGRP using the in vivo model.
Footnotes
Acknowledgements
This work was supported by the Swedish Medical Research Council (grant no. 5958) and the King Oscar V and Queen Victoria Foundation Sweden, the Danish Medical Research Council and the Lundbeck foundation Denmark.