
Abstract
The role of prostanoids in nociception is well established. The headache-eliciting effects of prostaglandin E2 (PGE2) and its possible mechanisms have previously not been systematically studied in man. We hypothesized that infusion of PGE2 might induce headache and vasodilation of cranial vessels. PGE2 (0.40 μg kg−1 min−1) or saline was infused for 25 min into 11 healthy subjects in a cross-over, double-blind study. Headache intensity was scored on a verbal rating scale from 0 to 10. In addition, we recorded mean flow in the middle cerebral artery (VMCA) by transcranial Doppler and diameter of the superficial temporal artery (STA) by high-resolution ultrasonography. All 11 subjects reported headache on the PGE2 day and no subjects reported headache on the placebo day (P = 0.001). During the immediate phase (0–30 min) (P = 0.005) and the postinfusion phase (30–90 min) (P = 0.005), the area under the curve for headache score was significantly larger on the PGE2 day compared with the placebo day. PGE2 caused dilatation of the STA (23.5%; 95% CI 14.0, 37.8) and the MCA (8.3%; 95% CI 4.0, 12.6). We suggest that PGE2 induces headache by activation and sensitization of cranial perivascular sensory afferents.
Introduction
Neurovascular headaches may originate from activation and sensitization of cranial perivascular afferents (1). The throbbing nature of migraine pain, head pain and corresponding arterial dilation after glyceryl trinitrate (2, 3), effect of arterial compression and dilation of arteries reported during attacks (4, 5) and animal models of migraine (1) suggest that vascular input may play an important role in generation of headache. However, it is still a matter of intense debate to what extent the vascular input contributes to head pain. Animal models suggest that mechanical dilation is not sufficient to sensitize perivascular afferents (6) and that release of inflammatory mediators by mast cell degranulation may play a key role in generation of head pain (7). The present human model has proven effective in the development of new principles for the treatment of migraine (2, 8–10).
Prostanoids such as prostaglandin E2 (PGE2) have been implicated in various experimental and clinical pain conditions (11). PGE2 is produced upon stimulation in cerebral endothelial cells (12) and mast cells (13). PGE2 released from endothelial cells causes strong dilation of cerebral vessels (14, 15). Furthermore, chemical and electrical stimulation of the trigeminal ganglion releases PGE2 from the dura mater (16), and PGE2 has been shown to sensitize sensory afferents (17). These data suggest that PGE2 may be involved in the pathophysiology of neurovascular headache.
Early open studies reported that PGE2 might cause a migraine-like pain in humans (18). However, the headache-eliciting effects of PGE2 have not been studied in a double-blind fashion, and data in animal models cannot be directly extrapolated to humans. We hypothesized that PGE2 may induce headache in healthy subjects accompanied by dilatation of intra- and extracranial arteries. Therefore, we aimed to study the headache-eliciting effect of PGE2 and to correlate any headache to the effect of PGE2 on cerebral and extracerebral arteries and cerebral blood flow (CBF) in healthy subjects in a double-blind, crossover study.
Design and methods
Pilot experiment
Before the main experiment, we conducted an open pilot study to find the optimal dose of PGE2 that caused a mild to moderate headache on a verbal rating scale (VRS) from 0 to 10 (0, no headache and 10, worst imaginable headache), without intolerable adverse effects and with detectable changes in mean blood flow velocity of the middle cerebral artery (VMCA) by transcranial Doppler (TCD). Three subjects (two male and one female) received intravenous PGE2 in stepwise increasing doses of 0.20, 0.40 and 0.60 μg kg−1 min−1. Each infusion lasted 25 min followed by a 60-min wash-out. The results of the pilot study showed that 0.40 μg kg−1 min−1 of PGE2 induced a mild to moderate headache in three subjects (VRS, range 1–3), was well tolerated and caused a 13.4% decrease in VMCA.
Main experiment
We recruited 14 healthy subjects (eight male and six female), mean age 22.1 years (range 19–26 years). Exclusion criteria were: a history of migraine or any other type of headache (except episodic tension-type headache less than once a month); any daily medication apart from oral contraceptives; serious somatic or psychiatric diseases. Subject 9 had a family history of migraine. The study was approved by the Ethics Committee of the County of Copenhagen (H-KA-20060026), Danish Medicines Agency, and the Danish Data Protection Agency and was undertaken in accordance with the Helsinki Declaration of 1964, as revised in Edinburgh in 2000. The study was registered on http://www.clinicaltrials.gov and monitored by the Good Clinical Practice unit at Copenhagen University Hospital. All subjects gave informed consent to participate.
Experimental design
In a double-blind, placebo-controlled, crossover design, the subjects were randomly allocated to receive PGE2 0.40 μg kg−1 min−1 or placebo (isotonic saline) over 25 min on 2 days, separated by at least a week. The central pharmacy performed the randomization and prepared the study drug. The randomization code remained in the hospital during the study and was not available to the investigators until the study was complete.
All subjects reported to the laboratory at 08.30 h headache free. Coffee, tea, cocoa or other methylxanthine-containing foods or beverages were not allowed for at least 8 h before start of the study. All procedures were performed in a quiet room, and room temperature was between 20.8 and 24.8°C. Subjects were placed in the supine position and a venous catheter (Venflon®) was inserted into the right antecubital vein for infusion. The subjects then rested for 30 min before baseline (0) recordings. After baseline measurements, infusion started using a time and volume controlled infusion pump (Braun Perfusor, Melsungen, Germany). Headache intensity, accompanying symptoms, VMCA, diameter of the superficial temporal artery (STA) and radial artery (RA), end-tidal partial pressure of pCO2 (PetCO2), adverse events and vital signs were recorded at baseline and then every 10 min until 90 min after start of infusion.
CBF measurements by single photon emission computed tomography (SPECT) were performed at baseline, 20 min and 60 min after start of infusion. Subjects were carefully instructed to complete a headache diary with accompanying symptoms according to the International Headache Society (19), including questions concerning premonitory symptoms (tired, yawning, stiff neck, blurred vision, thirst, intolerant/irritable, emotional, difficulty with concentration, any other symptoms) (20) and allodynia (combing, shaving, shower, cold, earrings, eyeglasses, heat, contact lenses, tight clothes, pillow, ponytail, necklace) (21) and any rescue medication every hour until 12 h after discharge from the hospital. Subjects were allowed to take rescue medication of their own choice at any time.
Headache intensity
Headache intensity was recorded on a VRS from 0 to 10 [0, no headache; 1, a very mild headache (including a feeling of pressing or throbbing—pre-pain); 5, moderate headache; 10, worst imaginable headache] (22).
Middle cerebral artery blood flow velocity
VMCA was recorded bilaterally by TCD with hand-held 2-MHz probes (Multidop X; DWL, Sipplingen, Germany), as previously described (23, 24). All recordings were done by the same skilled examiner (T.W.). PetCO2 (end-tidal CO2) was recorded simultaneously to the TCD measurements using an open mask that caused no respiratory resistance (ProPac Encore®; Welch Allyn Protocol, Beaverton, OR, USA). VMCA measurements were performed immediately after CBF measurements. Since regional cerebral blood flow (rCBFMCA) equals the product of VMCA and cross-sectional area of the MCA, assuming no change in cerebral blood volume, the relative percentage changes in MCA diameter (Δd) between treatments a and b were calculated as

Diameter of the superficial temporal artery and radial artery
Diameter of the frontal branch of the left STA and the left RA was measured by a high-resolution ultrasonography unit (20 MHz, bandwidth 15 MHz; Dermascan C; Cortex Technology, Hadsund, Denmark) as previously described (24, 26).
Cerebral blood flow
The examination was performed with the patient in the supine position, in quiet surroundings with eyes closed but ears unplugged. Four markers were drawn on the skin to ensure accurate positioning in each acquisition. PetCO2 was measured during each examination (Datex Normocap 200, Roedovre, Denmark). CBF was measured with 133Xe inhalation (Hevesy Laboratory, Ris⊘ National Laboratory, Denmark) and SPECT, using a brain-dedicated gamma camera (Ceraspect; DSI, Waltham, MA, USA). The system uses a stationary annular NaI crystal and a fast rotating collimator. Flow was calculated in each pixel based on the clearance curve, output was the ki value (27). To obtain CBF values, a partition coefficient (λ) of 0.85 was used. Calculation of flow in the perfusion territories of the major cerebral arteries was performed by fitting standard vascular regions of interest on axial slices of the brain as previously described (28). CBF values were corrected for significant changes in PetCO2 by 2% for each mmHg change in PetCO2(29). Six subjects were randomly allocated to CBF measurements.
Vital signs
Heart rate (HR) and blood pressure were measured every 10 min by an auto-inflatable cuff (ProPac Encore®; Welch Allyn Protocol). ECG (Cardiofax V; Nihon-Cohden, Japan) was monitored on an LCD screen and recorded on paper every 10 min.
Data analysis and statistics
Headache scores are presented as median and quartiles. Vascular baseline variables are presented as mean ±
The primary end-points were differences in the AUC for headache score (AUCheadache 0–30 min), AUCheadache 30–90 min, VMCA (AUCVMCA), STA (AUCSTA), RA (AUCRA) and PetCO2 (AUCPetCO2) between groups in the period 0–30 min. The secondary end-points were differences in HR (AUCHR), MAP (AUCMAP), SBP (AUCSBP) and DBP (AUCDBP) between groups in the period 0–30 min and 30–90 min. Additional explorative end-points were differences in AUC for all variables in the period 30–90 min. To test the differences between variables (including baseline variables) we used the Wilcoxon signed rank test for headache score and a paired, two-way t-test for vascular data.
gCBF, rCBFMCA and PetCO2 measured during CBF acquisition were also analysed for changes over time for each dose separately with univariate analysis of variance (
We tested for period and carry-over effects for all baseline variables with Mann–Whitney test (headache scores) and independent t-test (vascular variables). All analyses were performed with
Results
Eleven subjects (six male and five female, mean age 22.5 years, range 19–26 years) completed the study on both study days. One subject (1) was included, but later excluded before the first study day on suspicion of lack of compliance. Two subjects (6 and 13) were drop-outs on the second day (PGE2 day) due to adverse events. Subject 6 was withdrawn because of drop in MAP of 22% from baseline (security limits were ± 20% from baseline), dizziness, bilateral moderate throbbing headache (VRS 5) associated with phonophobia and aggravation by physical activity. Subject 13 wished to stop the experiment due to an urge to void, powerful low abdominal pain mimicking menstrual pain, and a unilateral mild throbbing headache (VRS 1) associated with nausea. These three subjects were excluded from statistical analysis. There were no differences in baseline recordings for any variables between the two experimental days (Table 1). There was no carry-over or period effect for baseline values of headache, VMCA, gCBF, rCBFMCA, STA, RA, MAP or HR (P > 0.05). We found no difference in baseline VMCA or rCBFMCA recordings between the left and right side on either active or placebo days (P > 0.05).
Mean baseline values (±

P-value, paired t-test.
Headache
During the observation period at hospital (0–90 min), 11 subjects reported headache on the PGE2 day and no subjects reported headache on the placebo day (P = 0.001, McNemar test) (Table 2). During the immediate phase, the AUCheadache 0–30 min on the PGE2 day, 25 (5–70), was significantly larger than on the placebo day, 0 (0–0), (P = 0.005) (Fig. 1). During the postinfusion phase, the AUCheadache 30–90 min on the PGE2 day, 35 (20–90), was significantly larger than on the placebo day, 0 (0–0), (P = 0.005) (Fig. 1). The median peak headache, 2 (0–2), occurred 40 min after start of PGE2 infusion.
Clinical characteristics of PGE2-induced headache in 11 healthy subjects
a, Peak headache on PGE2 day and associated symptoms during peak headache, in-hospital phase (0–90 min).
b, Peak headache on PGE2 day and associated symptoms during peak headache, out-of-hospital phase (1.5–14 h).
NS, not stated.

Median (□), individual active (▪) and placebo (x) headache scores on a verbal rating scale (VRS). There was a significantly higher headache response in the immediate (AUCheadache 0–30 min, P = 0.005) and postinfusion (AUCheadache 30–90 min, P = 0.005) phases on prostaglandin E2 day compared with placebo day.
During the delayed phase (1.5–14 h), five subjects reported headache on the PGE2 day, and three subjects reported delayed headache on the placebo day (P = 0.625, McNemar test) (Table 2). There was no difference in AUCheadache 30–90 min between the PGE2 day, 0 (0–2.50), and the placebo day, 0 (0–0.50) (P = 0.596).
Comparing all individual peak headaches, the peak immediate headache 2 (1–3.5) during the immediate and postinfusion phases on PGE2 day in subjects reporting delayed headache was identical to subjects not reporting delayed headache.
Comparing all individual peak headaches, the peak median headache was 1 (1–3) in 10 subjects during the infusion phase on PGE2 day, and this headache was reported to be bilateral (100%), throbbing (30%), constant (100%) and aggravating (20%). During the postinfusion phase peak median headache was 2 (1–2) in 10 subjects, and this headache was reported to be bilateral (100%), throbbing (10%), constant (100%) and aggravating (30%). During the delayed phase peak median headache was 1 (0–2) in five subjects, and this headache was reported to be bilateral (80%), constant (80%) and aggravating (40%).
Middle cerebral artery velocity, diameter and regional cerebral blood flow
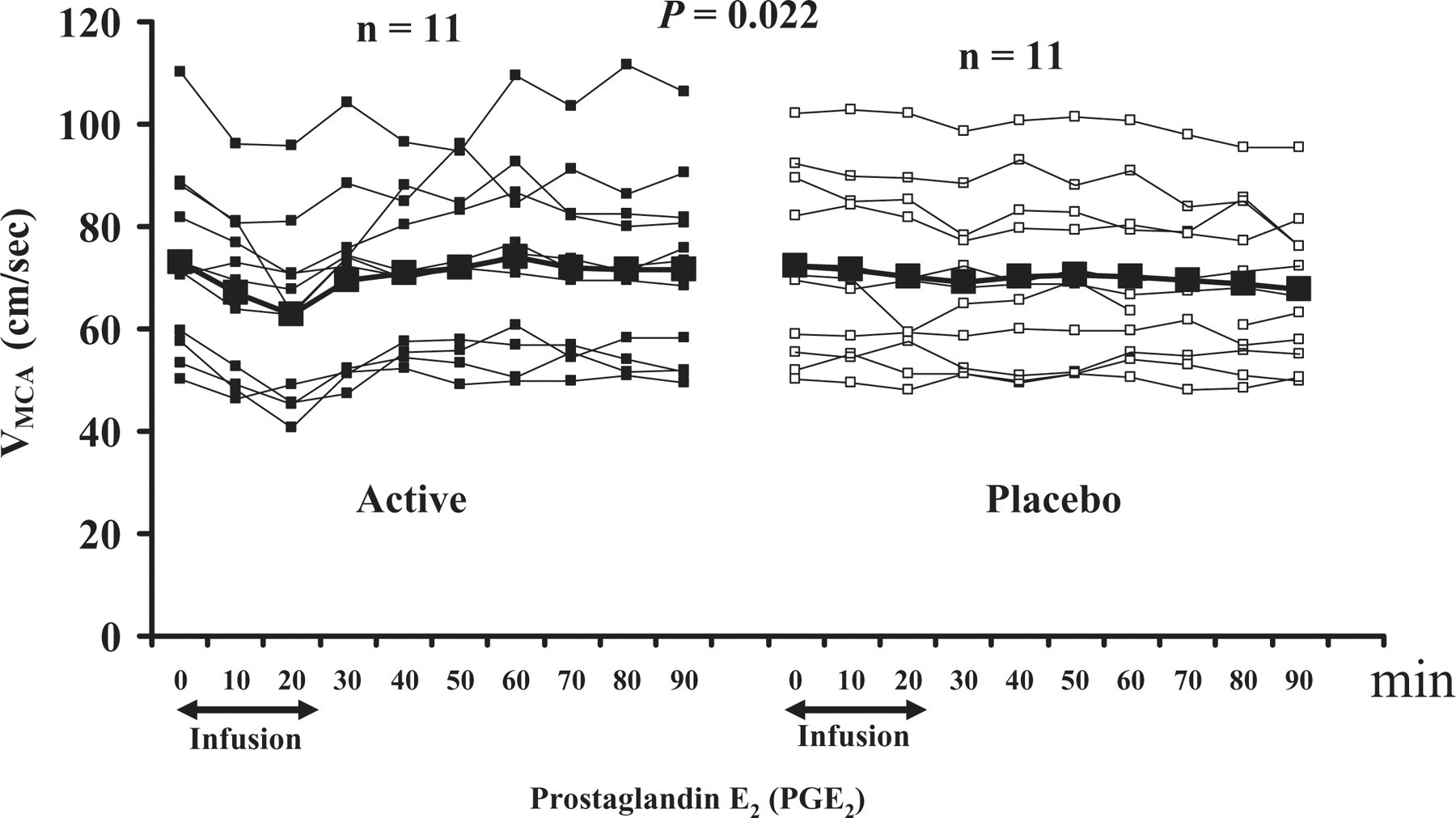
The AUCVMCA was significantly larger on PGE2 than on placebo during the immediate phase (P = 0.006). There was no difference in the AUCVMCA during the postinfusion phase (P = 0.362) (Fig. 2). There were no differences in the AUCPetCO2 recordings during TCD scans between PGE2 and placebo days, during either the immediate (P = 0.182) or postinfusion phases (P = 0.375).

Individual and mean flow velocities (cm/s) in the middle cerebral arteries (VMCA) on prostaglandin E2 compared with placebo day. There was a significant difference in AUCVMCA 0–30 min (P = 0.022).
There was no change in the AUC0–60 min for gCBF (P = 0.275) or for rCBFMCA (P = 0.221) after PGE2 compared with placebo. PetCO2 measured during CBF acquisition remained unchanged over time on the PGE2 day (P = 0.060), but decreased significantly on the placebo day (P = 0.025).
Maximal mean percentage changes from baseline (95% CI) in vascular variables on PGE2 day (0–90 min)

MCA, middle cerebral artery; STA, superficial temporal artery; RA, radial artery; HR, heart rate; MAP, mean arterial blood pressure; SBP, systolic blood pressure; DBP, diastolic blood pressure.
Superficial temporal artery
During the immediate phase, the AUCSTA 0–30 min on the PGE2 day was significantly larger than on the placebo day (P < 0.001) (Fig. 3). In the postinfusion period there was no difference in the AUCSTA 30–90 min on the PGE2 day compared with the placebo day (P = 0.845). The peak responses are presented in Table 3.
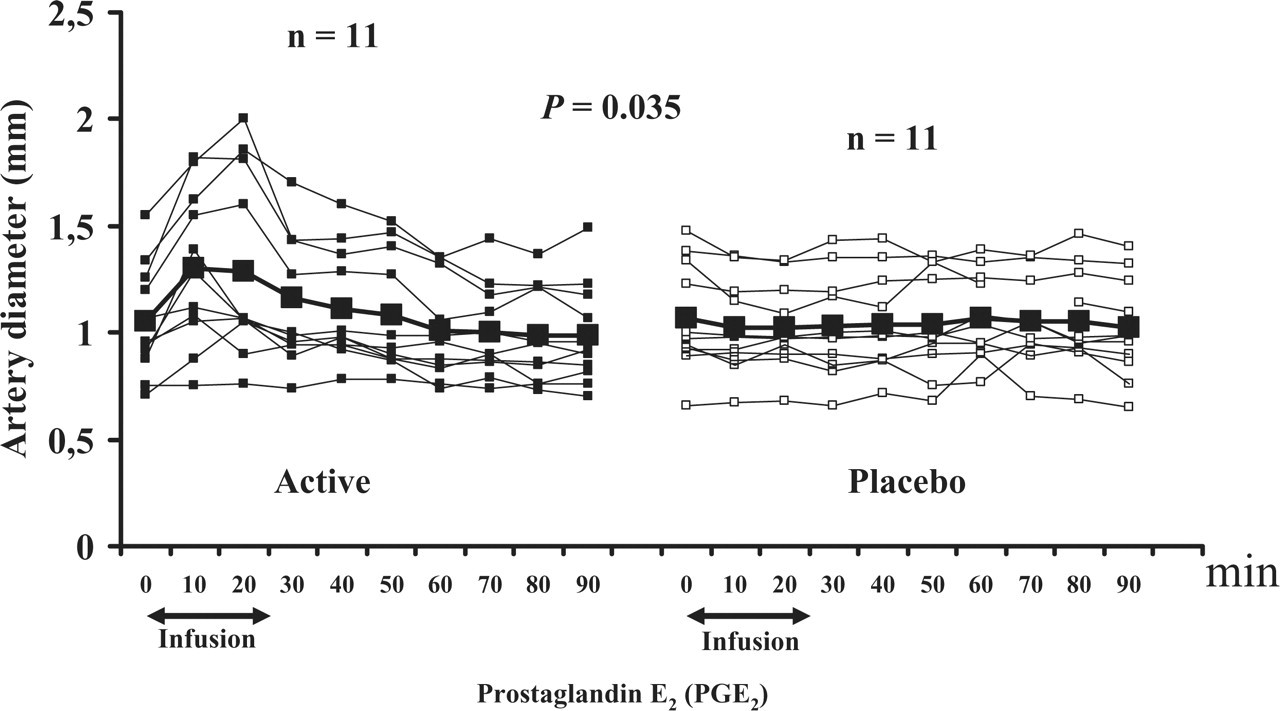
Individual and mean diameter (mm) of the superficial temporal artery (STA) on prostaglandin E2 compared with placebo day. There was a significant difference in the AUCSTA 0–30 min (P = 0.035).
Radial artery
There was no difference in the AUCRA 0–30 min (P = 0.856) between PGE2 day and placebo day. There was a significant difference in the AUCRA 30–90 min (P = 0.001) between PGE2 day and placebo day. The peak responses are shown in Table 3.
Allodynia and premonitory symptoms
There was no difference in premonitory symptoms between the two experimental days (P > 0.05, McNemar test). On the PGE2 day the following premonitory symptoms were reported: tiredness (91%), yawning (82%), thirst (55%), stiff neck (27%), intolerant (27%), emotional (18%), difficulty with concentration (18%), slept (9%) and heartbeat and dizziness (9%). On the placebo day, premonitory symptoms were reported as tiredness (55%), yawning (73%), stiff neck (27%), thirst (18%), difficulty with concentration (18%) and dizziness (9%).
There was no difference in reported allodynic symptoms between the two experimental days (P > 0.05, McNemar test).
Arterial blood pressure and heart rate
We found no difference in MAP or SBP between PGE2 and placebo days during either the immediate (AUCMAP 0–30 min, P = 0.343 and AUCSBP 0–30 min, P = 0.803) or the postinfusion phase (AUCMAP 30–90 min, P = 0.576 and AUCSBP 30–90 min, P = 0.066). There was a significant drop in DBP in the immediate phase (P = 0.002), but no difference in the postinfusion phase (P = 0.494) (Table 3).
AUCHR was significantly larger on PGE2 compared with placebo during the immediate (P < 0.001) and postinfusion phase (P < 0.001). The peak responses are shown in Table 3.
Adverse events
Adverse events were recorded and reported during immediate and postinfusion phases (Table 4). All subjects reported adverse events on the PGE2 day and three subjects reported adverse events on the placebo day.
Adverse events were recorded and reported during 0–90 min period
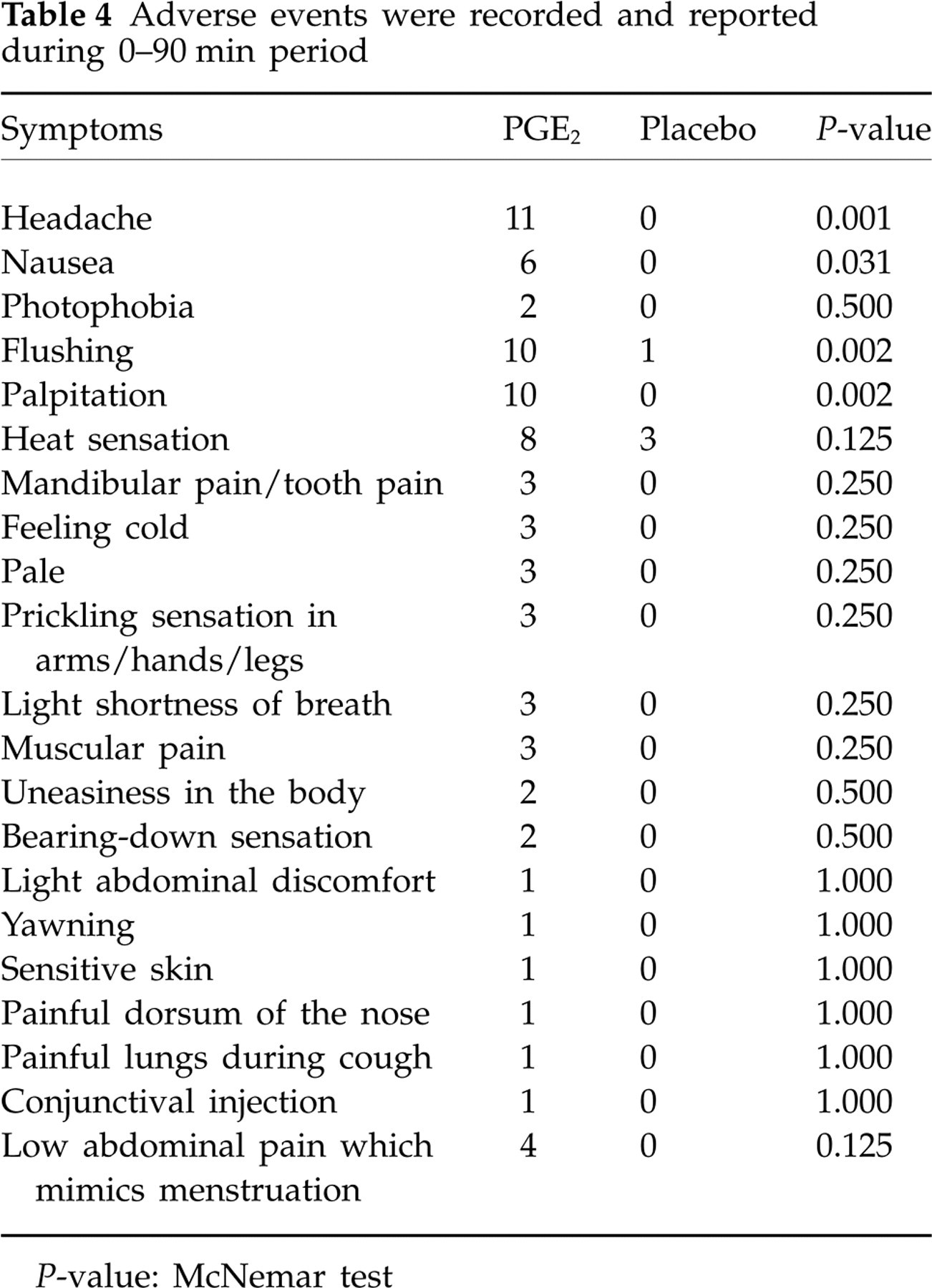
P-value: McNemar test
Discussion
To our knowledge, this is the first human double-blind study to explore a headache-eliciting and vasoactive effect of PGE2. The major outcome of the present study was that PGE2 induced extra- and intracranial vasodilation and a mild to moderate headache in healthy subjects. The present study cannot show the precise mechanism of PGE2-induced headache. Additional studies in both humans and animals are needed to show the underlying mechanisms, but we will discuss possible mechanisms of PGE2-induced headache.
Vascular mechanisms of PGE2-induced headache
PGE2 is a strong vasodilator in animal and human in vitro studies. Vasomotor actions of PGE2 depend on the type of tissue and type of receptor coupling (32). PGE2 dilates human MCA in vitro via the EP4-receptor (15). The EP2- and EP4-receptors are associated with smooth muscle relaxation. They are coupled to Gs-receptors causing elevation of intracellular cyclic adenosine 3′, 5′ monophosphate (cAMP) (33). In contrast to prostaglandin I2 (PGI2), it appears that dilation by PGE2 may involve potassium channels (34). Thus, KATP and Kca channels are involved in PGE2-induced pial artery dilation via cAMP and cyclic guanosine monophosphate pathways (35). Both these pathways have been implicated in the pathophysiology of neurovascular headaches (36, 37).
We have previously explored the role of the vasoactive prostanoid PGI2 in the generation of headache (38). The number of subjects (n = 11) who reported headache after PGI2 was the same as in the present study. The median peak headache intensity was slightly higher in the present study (2 vs. 1 on VRS). In contrast to PGI2, we observed a contraction of RA at 30 min after an initial dilation (10 min) (Table 3 and Fig. 4), but no contraction of MCA or STA. It has been reported that PGE2 given intravenously, intracarotidly and topically on cerebral arteries mediates both contraction and dilation of the arteries (39). Contraction has been shown in animal basilar and middle cerebral arteries with PGE2 in high doses (40). Different vasoactive responses to the same dose of PGE2 in cranial compared with peripheral arteries may reflect different functional roles of PGE2 in order to ensure sufficient blood flow to more vital organs.

Median headache (▪) and mean percentage changes from baseline for the middle cerebral artery blood flow velocity (VMCA) (○), diameter of the superficial temporal artery (STA) (▴) and diameter of the radial artery (RA) (•) on prostaglandin E2 day. Peak decrease in VMCA was 13.9% at 20 min compared with baseline. Peak increase in STA diameter was 23.5% at 10 min compared with baseline. Peak increase in RA was 4.4% at 10 min compared with baseline and peak decrease in RA was 10.0% at 30 min compared with baseline.
The magnitude of MCA (6.6%) and STA (38.8%) dilation during PGI2 was similar to dilation in the present study. Vasodilation by PGE2 was followed by a mild to moderate headache that came somewhat later (40 min after start of infusion) (Fig. 4). This suggests that other mechanisms rather than simple vasodilation are involved in the generation of headache. Mechanical dilation of the blood vessels is not sufficient to sensitize perivascular nociceptors (6). Furthermore, PGE2 increases permeability of the arterial wall (41) and is therefore able to reach perivascular afferents and sensitize them directly. In the following we will discuss a possible neuronal origin of PGE2-induced headache.
Neuronal mechanisms of PGE2-induced headache
The role of prostanoids in nociception is well established. The EP1, EP3 and EP4-receptors associated with PGE2 are expressed in the dorsal root ganglion neurons (42, 43). A chemical mixture containing various mediators including PGE2 activates and sensitizes dural nociceptors located on the meningeal vessel wall (6, 44), and infusion of cyclooxygenase (COX)-1/COX-2 inhibitors blocks sensitization in meningeal nociceptors (45). Meningeal mast cells may release inflammatory mediators including PGE2(13). Conversely, PGE2 can cause mast cell degranulation that may be suppressed by inhibitors of cAMP via protein kinase A (46). PGE2 releases calcitonin gene-related peptide (CGRP) (47) in primary sensory neurons. This is particularly relevant because administration of CGRP triggers migraine-like headache (9). Furthermore, it has been shown that PGE2 increases tetrodotoxin-resistant Na+ current (TTX-R INa) in nociceptors (48–50). The TTX-R INa channels are expressed in mechanosensitive meningeal nociceptors (51, 52), and these channels are involved in both the development and maintenance of acute and chronic inflammatory hyperalgesia (53). Furthermore, PGE2 increases or sensitizes the transient receptor potential vanilloid type 1 (TRPV1) responses in dorsal root ganglion neurons (54). This would activate TRPV1 and thereby cause spontaneous pain (54). Collectively, these data indicate that PGE2 in the present study may activate and sensitize meningeal nociceptors and thereby cause head pain.
The question is, does the action of PGE2 in the central nervous system (CNS) contribute to mechanisms of head pain? The EP2 receptor mRNA is expressed in the dorsal horn of the spinal cord (55), and PGE2 binding sites have been found in the spinal trigeminal nucleus and lamina I and II of the dorsal horn of the spinal cord (56). Peripheral inflammation up-regulates COX-2 mRNA in the spinal cord (57, 58) and increases production of prostanoids including PGE2. It has been shown that PGE2 releases CGRP (59) and nitric oxide (60) in the spinal cord. Furthermore, PGE2 passes the blood–brain–barrier (61) in a dose-dependent fashion (62) and might have a longer half-life in the CNS compared with plasma-like other prostanoids (62). PGE2 relieves dorsal horn nociceptive neurons from the inhibitory control by glycinergic neurons (63) via activation of EP2-receptors and a subsequent protein kinase A-dependent block of glycine receptors containing the α3 subunit (64). In animals, allodynia and hyperalgesia have been reported after intrathecal administration of PGE2 via EP1 and EP2 receptors (63, 65, 66). Thus, it is plausible to suggest that central mechanisms contribute to the headache reported in the present study.
Concluding remarks
The present study has shown that PGE2 dilates cephalic arteries in man and causes headache in healthy subjects. The prominent adverse events on the PGE2 day may have compromised blindness of the present study. However, the adverse events were caused by the physiological response to PGE2 and could not be avoided. The present double-blind approach is the best possible way of coping with methodological error. We suggest that PGE2-induced headache is caused by activation and sensitization of meningeal nociceptors. It would be relevant to study a possible migraine-inducing effect of PGE2.
Footnotes
Acknowledgements
The authors thank lab technicians Lene Elkjær, Annette Foldager and study nurse Kirsten Crawfurd for their dedicated and excellent assistance. The study was supported by grants from the Lundbeck Centre of Neurovascular Signalling (LUCENS) and the Danish Headache Society.