
Abstract
The vascular hypothesis of migraine has now been superseded by a more integrated theory that involves both vascular and neuronal components. It has been demonstrated that the visual aura experienced by some migraineurs arises from cortical spreading depression, and that this neuronal event may also activate perivascular nerve afferents, leading to vasodilation and neurogenic inflammation of the meningeal blood vessels and, thus, throbbing pain. The involvement of the parasympathetic system supplying the meninges also causes increased vasodilation and pain. As an acute attack progresses, sensory neurones in the trigeminal nucleus caudalis become sensitized, resulting in the phenomenon of cutaneous allodynia. Triptans may act at several points during the progression of a migraine attack. However, the development of central sensitization impacts upon the effectiveness of triptan therapy.
Introduction
The ‘classical’ or ‘vascular’ hypothesis of migraine developed by Wolff in the 1930s and 1940s proposed that migraine was simply a vasospastic disorder, initiated by vasoconstriction in the cranial vasculature (1). According to this theory, following the early vasoconstrictive stage, meningeal blood vessels dilate, activating the trigeminal sensory nerves that surround them, causing pain. This activation of trigeminal nerves also causes the release of vasoactive neuropeptides that further contribute to dilation, neurogenic inflammation of pain-sensitive cranial structures and, hence, worsening pain (2).
However, vasodilation alone cannot explain the local swelling and tenderness of the head that generally accompany migraine. A more integrated neurovascular theory has since been proposed, in which vascular change is considered secondary to neural activation. In addition, speculation continues over the role of primary central pain and the pain modulating system in migraine pathophysiology.
Understanding the pathophysiology of migraine is an important first step in learning how best to treat this condition. This article reviews the current understanding of migraine pathophysiology and discusses some of the more recent advances in this arena.
Cortical spreading depression
It was the Brazilian physiologist Leão, in the 1940s, who first showed that the application of potassium chloride directly on to the exposed cerebral cortex of the rat depressed electrical activity at the point of application and that over time this depression spread in a wave to adjacent areas of the cortex (3,4). This phenomenon is now known as the cortical spreading depression (CSD) of Leão. The electrical depression was associated with a reduction in cerebral blood flow.
A feline model, using diffusion-weighted magnetic resonance imaging (MRI), has been developed to investigate CSD in a gyrencephalic brain. Similar to the rat, primary waves of CSD were propagated across the cortex from the point of potassium application at a rate of 3–4 mm/min and were confined to the hemisphere in which they were initiated (5). Other experiments have shown that CSD is actually preceded by a brief phase of cortical spreading excitation (6).
In humans, a similar phenomenon of cortical reactivity is seen with the visual symptoms of aura that precede headache in some patients. Working at a similar time as Leão, Lashley had proposed that the growth of his own migrainous fortification spectrum (visual aura) corresponded to a disturbance consisting of a region of depressed neural activity bordered by a region of intense cortical excitation, proceeding across the occipital cortex at a rate of 2–3 mm/min (7).
More recently, Hadjikhani et al. demonstrated a direct association between CSD and aura in humans. In an experiment, migraineurs with visual aura were studied using functional MRI and blood oxygenation level dependent (BOLD) signal changes, which are a surrogate for CSD. At the onset of visual aura, a focal increase in BOLD signal (possibly reflecting vasodilation) developed within the extrastriate cortex and then progressed over the occipital cortex at a rate of 3–5 mm/min. The BOLD signal then diminished (possibly reflecting vasoconstriction after the initial vasodilation), with a corresponding subsequent decrease in visual activity, suggesting that an electrophysiological event such as CSD generates migraine aura in the human visual cortex (8). This correlates with the characteristic movement of visual symptoms from central to peripheral vision over 5–20 min, followed by the progression of a blind region (8).
More direct evidence that CSD underlies visual aura has been obtained with magnetoencephalography. Slow changes of the cortical magnetic field, corresponding to the potential changes that are produced by the neuronal depolarization in CSD, were observed in patients experiencing visual aura (9).
These demonstrations that cerebrovascular and magnetic field changes in migraineurs correlate with CSD supports the hypothesis that visual aura arises from CSD. Further support of the link between aura, electrical activity and blood flow can be drawn from the finding that the rate of progression of cortical spreading depression is the same as that of both oligemia and migrainous scotoma.
Neurovascular mechanisms
Migraine is now considered to be a neurovascular pain syndrome in which there is abnormal neuronal excitability in the cerebral cortex, possibly due to reduced levels of magnesium and increased levels of calcium and glutamate, producing susceptibility to CSD in migraineurs, and peripheral sensitization of the trigeminal vascular system. Pain is generated from the dura and blood vessels and is enhanced by neurogenic plasma protein extravasation, which is mediated by neuropeptides (10–12). In addition, central neural processing occurs through mechanisms of pain modulation and central sensitization (Fig. 1).
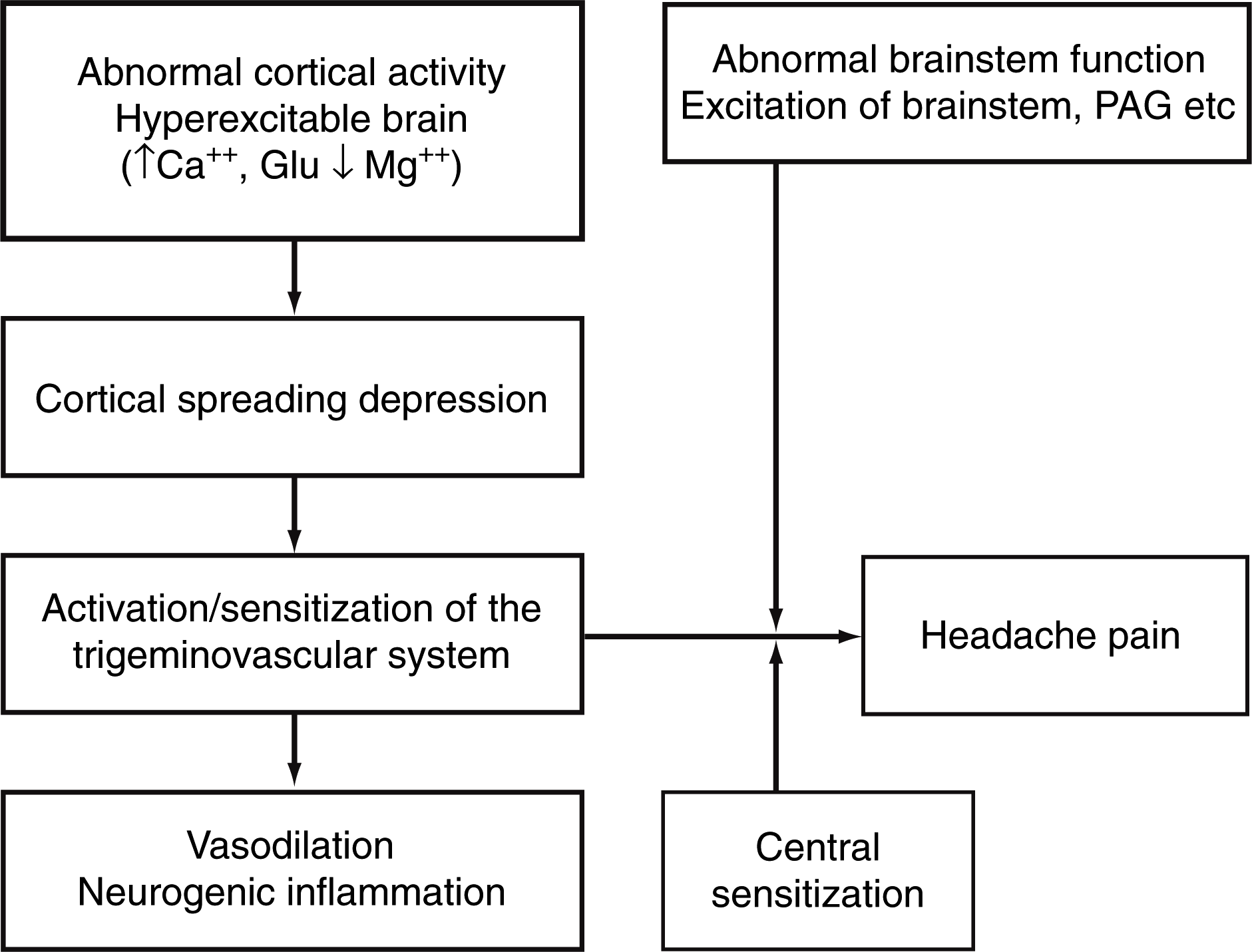
Proposed mechanisms of migraine headache. Reprinted from (13) with permission. © 2003, MacMillan Magazines Ltd.
Each trigeminal ganglion cell is known to project single axons to the pia arachnoid and its blood vessels and to the dura mater. Its central projections lie in the trigeminal nucleus caudalis (TNc). The presynaptic terminals of the trigeminal nerves contain the neuropeptides substance P, calcitonin gene-related peptide (CGRP), and neurokinin A (NKA). In addition, 5-HT1D/1F heteroreceptors are present on the trigeminal nerve ending, and stimulation of these inhibits the release of substance P and CGRP. Post-synaptically, blood vessels contain 5-HT1B receptors, which produce vasoconstriction when stimulated, and the central nervous system contains 5-HT1F/1B heteroreceptors; these are important as they inhibit the release of 5-HT, noradrenaline and acetylcholine.
Depolarization of the trigeminal sensory C-fibres innervating the cerebral blood vessels therefore results in the release of neuropeptides, which interact with the blood vessel wall, causing dilation, plasma protein exudation, and the activation and degranulation of mast cells – the phenomenon of neurogenic inflammation. The exudation of plasma protein has been confirmed by Buzzi and Moskowitz, using iodine-125 labelled albumin in rats and guinea pigs. Administration of 5-HT or neuropeptides or direct electrical stimulation of the trigeminal nerves significantly increased albumin exudation compared with control animals (14).
In such models, both triptans and dihydroergotamine have been shown to block plasma extravasation in the dura following unilateral stimulation of the trigeminal nerves, but not that produced by administration of substance P or NKA (14). In addition, both drugs significantly decreased the number of activated mast cells and attenuated the increase in CGRP released following trigeminal stimulation (15–17).
The triptans, established as 5-HT1B/1D receptor agonists, may act at the inhibitory heteroreceptor on the presynaptic trigeminal nerve ending. They may also act postsynaptically at cerebral blood vessels and in the central nervous system. Understanding their mechanism of action may therefore help to elucidate pathophysiological pathways.
Moskowitz and coworkers have investigated the link between aura and headache. Aura occurs in the primary visual cortex, primary somatosensory cortex and primary motor cortex – grey matter structures in close proximity to the trigeminal vascular system innervating the surface of the brain. CSD releases potassium ions, protons, nitric oxide (NO), arachidonic acid, prostaglandins and amino acids from the pia arachnoid, in amounts sufficient to sensitize or activate perivascular nerve afferents. Information then travels both through an axon reflex to the dura, producing vasodilation and plasma protein extravasation, and also centrally through a pathway following the trigeminal ganglia to the TNc. The central route then follows a reflex arc to the parasympathetic outflow, which passes through the superior salivatory nucleus, the cranial ganglia and sphenopalatine ganglia before finally reaching the meninges, causing vasodilation and pain (Fig. 2) (13,19,20). Therefore, in addition to the mechanisms of cortical depression and extravasation, central modulation of migraine occurs, and drugs acting only on plasma extravasation (or neurogenic inflammation) will not halt the development of headache.
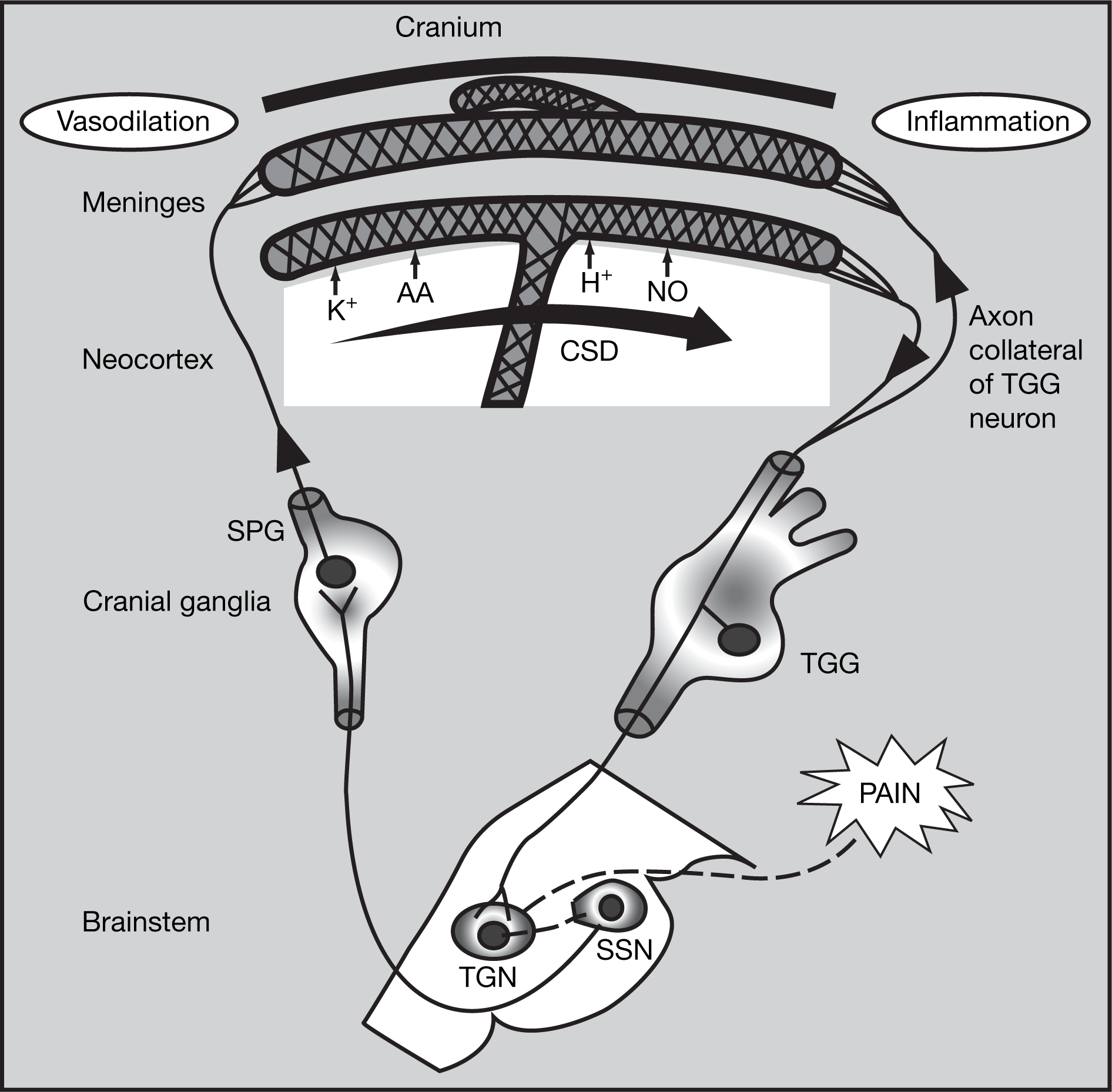
Key pathways in aura and headache. Reproduced from (18) with permission.
Role of the brainstem
The potential link between CSD and headache has now been identified, but the mechanism of central modulation of migraine is still unclear. Debate exists as to whether the brainstem might act as a generator or modulator of migraine. MRI studies have shown that the periaqueductal grey (PAG) is involved in pain modulation. In subjects paying attention to a painful stimulus, the PAG was not activated and the perceived pain intensity was high; when the same subjects were distracted from their pain, the PAG was activated and the pain intensity was decreased (21). Activation of the PAG therefore induces analgesia.
Weiller et al. have shown that aminergic nuclei (locus cereleus, raphe nuclei) in the brainstem modify trigeminal pain processing during migraine. Using PET, brainstem activation has been visualized during spontaneous migraine attacks, even after successful (i.e. pain-relieving) migraine treatment (22). Therefore, brainstem nuclei nociceptors that are involved in the central control of pain may be dysfunctional in migraineurs, and may have an increased tolerance for trigeminal neuronal hyperexcitability (13).
Peripheral sensitization of trigeminal neurones to mechanical or chemical stimuli mediates the throbbing pain of migraine, and its worsening after coughing or other normally innocuous activities that increase intracranial pressure. Central sensitization of the second-order TNc neurones receiving sensory input from the dura and the skin results in cutaneous allodynia (13,23,24).
It has been demonstrated that the mechanism of central sensitization involves wide dynamic range (WDR) neurones in the TNc, which normally receive input from C-fibres in the trigeminal ganglia. Following peripheral sensitization and neurogenic inflammation, increases in the concentrations of mediators such as 5-HT, glutamate, substance P, CGRP, NO and protons act to increase the input from the C-fibres to the WDR neurones, resulting in the WDR neurones modifying their responses to the input they receive from Aβ fibres (central sensitization). Under normal circumstances, the input from Aβ fibres is not painful. During allodynia, the sensitized WDR neurones respond to the input to generate a pain response (25,26).
It is therefore thought that migraine develops as a result of changes in the processing of pain and sensory input. Aura is triggered in the hypersensitive cortex, and headache is probably generated by central pain facilitation in combination with neurogenic inflammation, as well as peripheral activation and sensitization. Brainstem nociceptor sensitization may occur prior to or simultaneously with the development of neurogenic inflammation.
Recent advances in understanding
Oshinsky has looked more closely at the sensitization of brainstem WDR neurones in rats. Lamina V second-order sensory neurones in the TNc with periorbital receptive fields were isolated (27), and an ‘inflammatory soup’– containing histamine, 5-HT, bradykinin and prostaglandin E2 at physiological pH (25,28) was applied to the dura, and receptive fields and response thresholds were measured by brushing with von Frey hairs before and at fixed times after application of the inflammatory soup. The inflammatory soup produced a significant increase in the intensity of neuronal activity, i.e. sensitization, in response to brushing with the von Frey hairs at 2 h after application, compared with that observed prior to application and also when compared with an application of saline. Similar results were obtained when the von Frey hairs were used to exert gentle pressure on the dura before and after application of the inflammatory soup, i.e. a dramatic change in pressure response threshold. In addition, the administration of triptans prior to application of the inflammatory soup blocked the development of sensitization in this model (29). This model may therefore be used to test novel acute and preventive migraine medications.
An extension of these rat studies has utilized microdialysis with an inflammatory soup in the perfusate. These have revealed that an initial decrease in extracellular glutamate followed by a large increase in extracellular glutamate occurs after perfusion of the inflammatory soup, compared with saline. The time course of the increase in glutamate corresponds to the change in pressure threshold from the earlier studies, confirming a role for glutamate in sensitization (30,31).
Oshinsky has proposed that the delayed sensitization (i.e. the delay between applying the inflammatory soup and the increase in extracellular glutamate) may be due to descending control through serotonergic and noradrenergic systems, and that this may be the basic mechanism of upper brainstem control of the TNc. It has therefore also been proposed that the triptans may work via this mechanism (29).
Clinical implications: a rationale for early triptan therapy
During the progression of an acute migraine attack, when the peripheral pain fibres become sensitized, the pain begins to throb and is worsened by movement. Cutaneous allodynia develops in around 80% of migraineurs in the later stages of an attack, when central sensitization of the second-order sensory TNc neurones occurs. Under these circumstances, sufferers may be unable to comb their hair, put on their glasses or perform any other action that requires touching the skin of the face or scalp (24). In addition, the third-order sensory neurones in the thalamus may then also become sensitized, leading to the extracephalic allodynia experienced by some migraineurs (25). When taken at this late stage of an attack, triptans may be less effective than when taken earlier, i.e. prior to central sensitization (32).
Early triptan treatment, up to 60 min after the onset of pain and before cutaneous allodynia is established, is particularly effective in relieving headache pain during a migraine attack. In a recent clinical trial, 93% of patients with migraine but no allodynia treated with a triptan were pain-free at 2 h compared with only 15% of patients with migraine experiencing allodynia (33).
Therefore, it has been proposed that during a migraine attack, peripheral sensitization occurs and central neurones are initially activated (but not yet sensitized), with the occurrence of pain and throbbing. If a triptan is administered at this stage, central sensitization may still occur but the release of neuropeptides is blocked from both the peripheral and central nerve endings, and these neurones become quiescent, the pain and throbbing is relieved and allodynia does not develop. However, if triptan therapy is not initiated until after central sensitization has occurred, the throbbing is relieved, but the pain and allodynia remain. This is the clinical rationale for early treatment with a triptan during a migraine attack.
Summary
Understanding the pathophysiology of migraine, including an understanding of early and late-phase mechanisms (in particular neuronal hyperexcitability, neurogenic inflammation and peripheral and central sensitization), will help clinicians to manage migraine more effectively. It will help them address the progression of an attack, as well as giving them the tools in acute management that are necessary to combat migraine-associated disability, socioeconomic impact and progression to chronic migraine. In particular, recognizing the clinical importance of the development of cutaneous allodynia in triptan treatment response can produce improved patient outcomes.
Triptans may act at several different points during the progression of a migraine attack, including inhibiting neurogenic inflammation, as vasoconstrictors of the cerebral blood vessels and also by blocking the transmission of pain from the peripheral trigeminal nerves to the brainstem, where central sensitization occurs.