
Abstract
The authors investigated the effect of vascular endothelial growth factor (VEGF) on hypoxic injury of cultured rat hippocampal neurons. Treatment with glutamate receptor antagonists prevented hypoxic neuron death. The same magnitude of protection was observed in cultures treated with VEGF, which also reduced excitotoxic neuron death induced directly by an exposure to N-methyl-
Vascular endothelial growth factor (VEGF) promotes physiological and pathological angiogenesis (Carmeliet and Collen, 2000; Plate, 1999). Expression of VEGF is activated by the transcription factor hypoxia-inducible factor-1α (Carmeliet and Collen, 2000). In the nervous system, VEGF is induced in response to hypoxia/ischemia in neurons, microglia/macrophages, and endothelial cells (Hayashi et al., 1997; Lennmyr et al., 1998; Plate et al., 1999). Mice carrying a deletion of the hypoxia-response element in the VEGF promoter showed normal angiogenesis, but a degeneration of motoneurons, suggesting that VEGF may also exert neuroprotective effects (Oosthuyse et al., 2001). Other studies have shown that VEGF is also able to stimulate axonal outgrowth (Sondell et al., 2000) and neurogenesis (Yang and Cepko, 1996; Yourey et al., 2000). These effects are mediated via the VEGF-receptor 2 (Flk-1) and its coreceptor neuropilin-1 (NRP-1), which are expressed constitutively in many neurons (Sondell et al., 2000; Yang and Cepko, 1996).
Topical or intravenous administration of VEGF has been shown to protect brain tissue against ischemic injury and to promote angiogenesis after ischemia (Hayashi et al., 1998; Zhang et al., 2000). However, VEGF may also enhance ischemic injury due to its ability to increase blood-brain barrier leakage, particularly during the early stage of an ischemic insult (van Bruggen et al., 1999; Zhang et al., 2000). It has previously been demonstrated that VEGF exerts intrinsic neuroprotective properties on primary central neurons during hypoxia (Jin et al., 2001). VEGF enhances the survival of non-neural and neural cell lines in response to hypoxia and serum deprivation in a PI-3-kinase-dependent manner by inhibiting the activation of caspases, a family of proapoptotic proteases (Baek et al., 2000; Jin et al., 2000). The present study demonstrates that VEGF protects cultured rat hippocampal neurons against hypoxic injury and demonstrates that antiexcitotoxic effects rather than inhibition of caspases are involved in this protection.
MATERIALS AND METHODS
Recombinant human VEGF165 (rhVEGF165), N-methyl-
Cell culture
Cultured hippocampal neurons were prepared from neonatal (P1) Wistar rats (Krohn et al., 1998). Cells were plated on poly-D-lysine-coated 6- or 24-well plates. All experiments were performed on 12- to 14-day-old cultures. Animal care followed official government guidelines. Human umbilical vein endothelial cells and rat pheochromocytoma PC12 cells were prepared and cultured as described elsewhere (Bui et al., 2001; Levkau et al., 1998).
Reverse transcription polymerase chain reaction and immunofluorescence analysis
Reverse transcription polymerase chain reaction (RT-PCR) and immunofluorescence analysis were performed as described previously (Bui et al., 2001). The sequences of the primers used were as follows: Flk-1 sense primer, 5'-TGCCTTTGGC-CAAGTGATTGA-3'; Flk-1 antisense primer, 5'-CCAGGTCCCTGTGGATACA-3'; Flt-1 sense primer, 5'-TATGCCTGCAGAGCCAGGAA-3'; Flt-1 antisense primer, 5'-AACTCCCACTTGCTGGCATC-3'; NRP-1 sense primer, 5'-CCTTGCTCTGGAATGTTGGG-3', NRP-1 antisense primer, 5'-TGGCTCTCTCGGGGTAGAT-3'. Primers were directed against identical regions in rat and human sequences. PCR was carried out for 35 cycles at 94°C for 30 seconds, 52°C for 30 seconds, and 72°C for 60 seconds. To rule out contamination of RNA preparations with genomic DNA, RT was inactivated at 95°C for 10 minutes prior to cDNA synthesis in control experiments. Subsequent PCR reaction revealed no amplification products. Flk-1 protein expression was detected with a mouse monoclonal antibody (sc-6251; Santa Cruz), diluted 1:20. Active calpain I was detected using a mouse monoclonal antibody specific for the large subunit of postautolytic calpain I, diluted 1:200 (Samis et al., 1987; kindly provided by Dr. J.S. Elce; Queen's University, Kingston, Canada). Chromatin was stained with Hoechst 33258.
Hypoxia
Cultures were transferred to a humidified hypoxic chamber (37°C, 95% nitrogen, 5% CO2, 0.2% O2; COY Laboratory Products). Controls were maintained under normoxic conditions.
NMDA-induced excitotoxicity
Cultures were washed in HEPES-buffered saline (HBS) and incubated for 5 minutes in Mg +-free HBS containing 300-μmol/L NMDA (Luetjens et al., 2000). Controls were exposed to Mg2+-free HBS. After the exposure cells were washed with HBS and returned to the original culture medium.
Staurosporine-induced apoptosis
Staurosporine was added directly into the culture medium to a final concentration of 100 nmol/L (Krohn et al., 1998). Controls received the vehicle (dimethylsulfoxide; 0.1% V/V).
Evaluation of cell death
Cell death was assessed by the trypan-blue dye exclusion method. A total of 300 to 500 neurons were counted in three to four randomized subfields of each culture. Two investigators performed the cell counts without knowledge of the respective treatment.
Electrophoretic mobility shift assay
Whole-cell extracts were homogenized in 100 μL TOTEX lysis buffer (20 mmol/L HEPES, 350 mmol/L NaCl, 1 mmol/L MgCl2, 0.5 mmol/L EDTA, 0.1 mmol/L EGTA, 20% glycerol, 1% Nonident P-40, 0.5 mmol/L DTT, 2 mmol/L PMSF, 50 μg/mL aprotinin, 10 μg/mL leupeptin; pH 7.9) and centrifugated at 13,000 U/min for 15 minutes The recognition sequence of the double stranded transcription factor nuclear factor-KB (NFκB) was end-labeled with [γ−32P]ATP using T4 polynucleotide kinase. The reaction mixture, containing 10 μg protein homogenate and 50,000-cpm labeled oligonucleotide in 26 μL incubation puffer (20 mmol/L HEPES, pH 7.5; 50 mmol/L KCl, 2.5 mmol/L MgCl2, 20% Ficoll, 1 mmol/L DTT, 2 μg poly[dI-dC], 2 μg bovine serum albumin) was incubated for 15 min at room temperature. Reaction mixtures were run on 4% polyacrylamide gels for 2 hours. Gels were exposed at −80 °C.
Caspase-3–like protease activity
Activation of caspase-3–like proteases was determined by measuring the cleavage of the fluorigenic substrate acetyl-DEVD-aminomethylcoumarin (Ac-DEVD-AMC) by cytosolic extracts (Krohn et al., 1998). The fluorigenic substrate is cleaved by effector caspase-3, 6, and 7, as well as by caspase-8 and 9 (Garcia-Calvo et al., 1999). Production of fluorescent AMC was monitored over 60 minutes. Caspase-3–like activity is expressed as change in fluorescence units per hour per microgram protein.
Statistics
Data are given as mean ± SD. For statistical comparison, one-way analysis of variance followed by Tukey test were employed. P values less than 0.05 were considered statistically significant.
RESULTS
The RT-PCR analysis demonstrated that cultured rat hippocampal neurons and the rat PC12 pheochromocytoma cell line expressed both Flk-1 and NRP-1 mRNA (Fig. 1A). In contrast, expression of Flt-1 mRNA could not be detected, but was present in endothelial cells that served as positive controls. Flk-1 protein was expressed in most hippocampal neurons as demonstrated by immunofluorescence analysis (Fig. 1B). Subsequently, 14-day-old hippocampal neuron cultures were subjected to hypoxia. rhVEGF165 was added at a concentration of 50 ng/mL immediately before the cultures were transferred into the hypoxic chamber. Evaluation of cell death after 24 and 48 hours demonstrated that VEGF potently protected against hypoxic neuronal injury (Fig. 1C). Hypoxic neuron death has been shown to involve glutamate release and subsequent excitotoxic injury (Choi, 1994). A similar degree of protection was observed in cultures treated with the NMDA receptor antagonist MK-801 (1 μmol/L) plus the AMPA receptor antagonist NBQX (10 μmol/L). A combined treatment with VEGF and the glutamate receptor antagonists did not result in an enhanced neuroprotection; instead a tendency towards increased cell death rates compared with the single treatments was evident (Fig. 1C). Activation of transcription factor NFκB has been implicated in the biological effects of VEGF in endothelial cells (Kim et al., 2001; Marumo et al., 1999). Hypoxia per se led to an activation of NFκB in the hippocampal cultures as evidenced by electrophoretic mobility shift assay (EMSA) analysis (Fig. 1D). Treatment with VEGF, however, did not significantly modify NFκB activation during hypoxia.
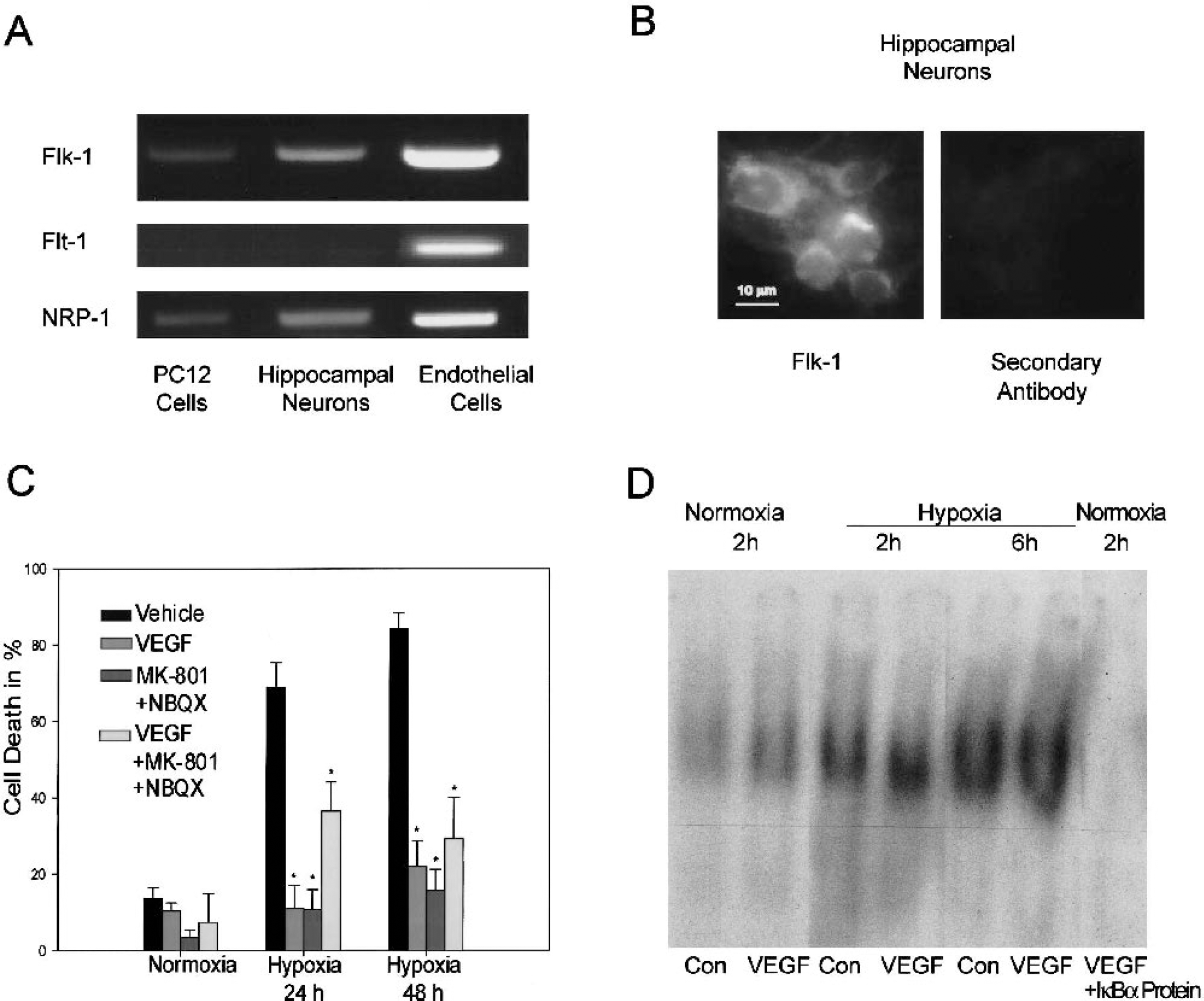
Vascular endothelial growth factor protects hippocampal neurons against hypoxic injury.
We subsequently investigated whether the neuroprotective activity of VEGF was associated with an inhibition of effector caspases during hypoxic neuron death. Hypoxic injury was not associated with the activation of DEVDases in the hippocampal neuron cultures (Fig. 2A). In contrast, we detected the activation of calpain I during hypoxia (Fig. 2B). A 4-hour pretreatment with VEGF also failed to protect rat hippocampal neurons against the activation of effector caspases induced by the proapoptotic agent STS (Fig. 2C).
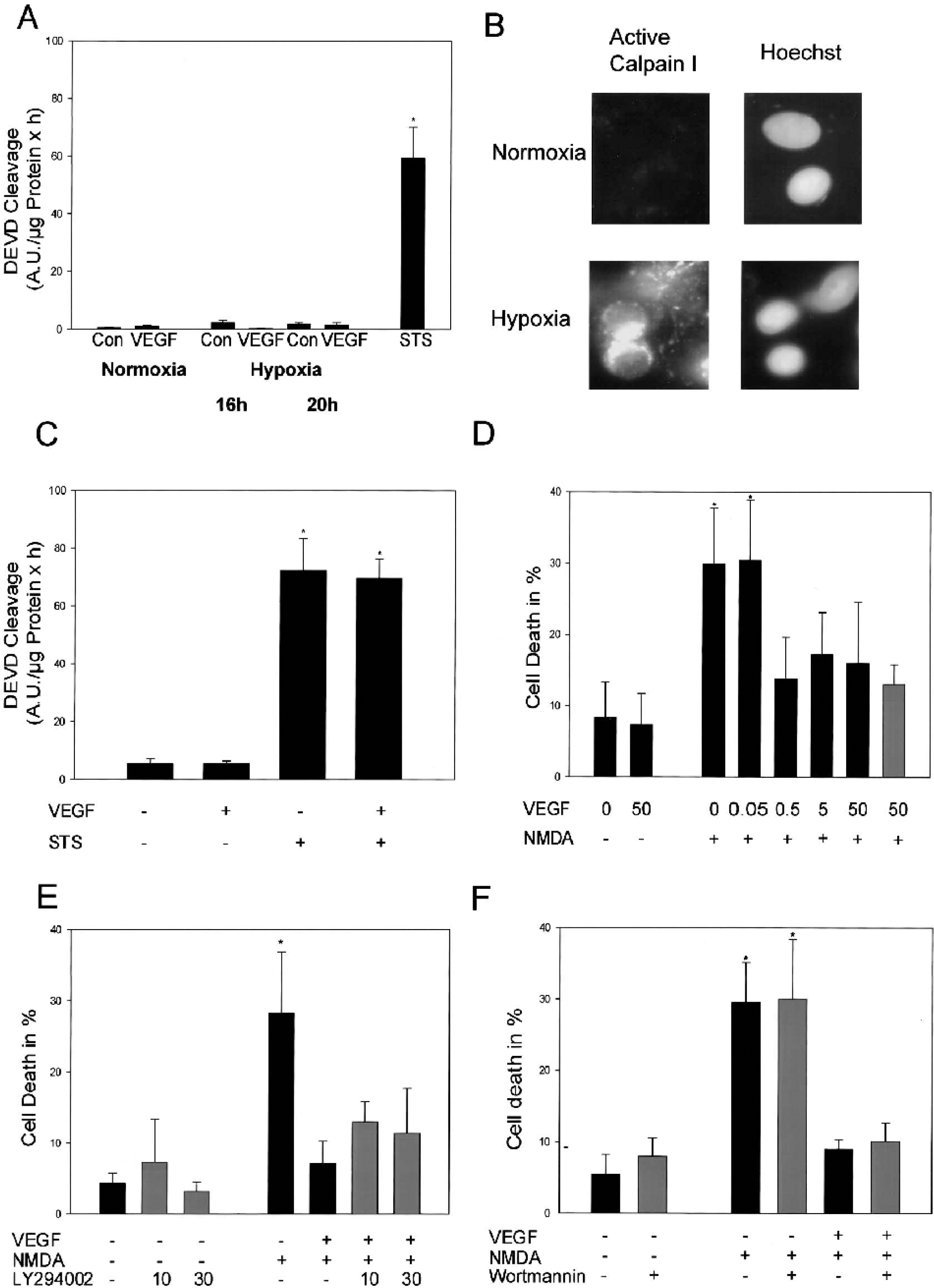
Mechanisms of VEGF-induced protection.
Interestingly, a 4-hour pretreatment with VEGF potently protected the hippocampal neurons against excitotoxic injury induced by an exposure to NMDA (Fig. 2D). Dose-response analyses of the protective effect of VEGF against excitotoxic (Fig. 2D) and hypoxic neuron death (data not shown) revealed that potent neuroprotective activities were notable at concentrations as low as 0.5 ng/mL. VEGF also protected against excitotoxic injury when added to the cultures only during and after the NMDA exposure (Fig. 2D). The antiexcitotoxic effect of VEGF was not significantly diminished in cells treated with the PI-3 kinase inhibitors LY294002 or wortmannin (Figs. 2E and 2F), suggesting that antiapoptotic signaling through PI-3 kinase was not involved in VEGF-induced neuroprotection.
DISCUSSION
The present study demonstrates that VEGF protects cultured rat hippocampal neurons against hypoxic injury via a direct antiexcitotoxic, caspase-independent mechanism. Both hypoxic injury and NMDA-induced excitotoxicity were associated with the activation of calpain I, but a significant activation of effector caspases could not be detected (Fig. 2; Lankiewicz et al., 2000). We have recently demonstrated that the lack of effector caspase activation during excitotoxic (and therefore also hypoxic) neuron death may be due to calpain-mediated degradation of executioner caspases (Lankiewicz et al., 2000). VEGF also failed to inhibit the activation of effector caspases in response to STS, a well-characterized proapoptotic agent (Krohn et al., 1998). Furthermore, the antiapoptotic PI-3 kinase pathway was not involved in VEGF-induced neuroprotection. Our data therefore suggest that the PI-3-kinase-dependent suppression of caspase-dependent cell death pathways—observed in cell lines lacking functional glutamate receptors (Baek et al., 2000; Jin et al., 2000)—as well as direct antiexcitotoxic effects both play a role in VEGF's protection against hypoxic neuronal injury. Interestingly, VEGF also failed to modify the activation of the antiapoptotic and proinflammatory transcription factor NFκB during hypoxia.
Our data suggest that signaling via the Flk-1 receptors pathway mediates the protective effect of VEGF. NRP-1 was expressed in the hippocampal neuron cultures and may act to prolong VEGF binding of Flk-1 receptors (Petrova et al., 1999). The protective effects of VEGF against NMDA-induced excitotoxic injury suggest that a hypoxia-dependent upregulation of VEGF signaling components is not required for a neuroprotective effect, and hence that constitutive expression of Flk-1 and NRP-1 is sufficient to mediate neuroprotection. Deletion of the hypoxia-response element in the VEGF promoter caused motor neuron degeneration in mice resembling amyotrophic lateral sclerosis (Osthuyse et al., 2001). It is possible that the lack of antiexcitotoxic effects of VEGF may trigger motoneuron degeneration in these mice. Excitotoxic mechanism have also been implicated in the pathogenesis of familial and sporadic amyotrophic lateral sclerosis (Howland et al., 2002; Leigh and Meldrum, 1996), suggesting that this neuron type is very susceptible to excitotoxic challenges and relies on VEGF as an endogenous neuroprotectant. The results reported here suggest that VEGF may also be an important survival factor for other central neurons by inhibiting potentially harmful, excitotoxic processes. In contrast to glutamate receptor antagonists, VEGF may also stimulate angiogenesis and neurogenesis. Therefore, VEGF and the VEGF-induced signaling cascade may become interesting targets for the treatment of cerebral hypoxia and ischemic stroke.
Footnotes
Acknowledgements
The authors thank Christiane Schettler for excellent technical assistance, and Dr. J. C. Elce for providing the calpain antibody used in this study.