
Abstract
Delayed administration of vascular endothelial growth factor (VEGF) promotes functional recovery after focal cerebral ischemia. However, early intravenous injection of VEGF increases blood–brain barrier (BBB) leakage, hemorrhagic transformation and infarct volume whereas its application to cortical surface is neuroprotective. We have investigated whether or not early intracerebroventricular administration of VEGF could replicate the neuroprotective effect observed with topical application and the mechanism of action of this protection. Mice were subjected to 90 mins middle cerebral artery (MCA) occlusion and 24 h of reperfusion. Vascular endothelial growth factor (8 ng, intracerebroventricular) was administered 1 or 3 h after reperfusion. Compared with the vehicle-treated (intracerebroventricular) group, VEGF decreased the infarct volume along with BBB leakage in both treatment groups. Neurologic disability scores improved in parallel to the changes in infarct volume. Independently of the decrease in infarct size, VEGF also reduced the number of TUNEL-positive apoptotic neurons. Phospo-Akt levels were significantly higher in ischemic hemispheres of the VEGF-treated mice. Contrary to intracerebroventricular route, intravenous administration of VEGF (15 μg/kg) enhanced the infarct volume as previously reported for the rat. In conclusion, single intracerebroventricular injection of VEGF protects brain against ischemia without adversely affecting BBB permeability, and has a relatively long therapeutic time window. This early neuroprotective action, observed well before recovery-promoting actions such as angiogenesis, possibly involves activation of the PI-3-Akt pathway.
Keywords
Introduction
Vascular endothelial growth factor (VEGF) is induced by hypoxia/ischemia (Brogi et al, 1994; Schoch et al, 2002; Bernaudin et al, 2002) and is thought to contribute to postinjury angiogenesis (Krupinski et al, 1994). Vascular endothelial growth factor is a 34 to 48 kDa homodimeric glycoprotein originally isolated as both an endothelial mitogen and a potent enhancer of vascular permeability (Conolly et al, 1989). Five isoforms generated by alternative splicing have been isolated in humans, and four isoforms have been detected in murines (Sondell et al, 1999). Vascular endothelial growth factor exerts its mitogenic action by binding to tyrosine kinase receptors, VEGFR-1 or Flt-1 and VEGFR-2 or Flk-1/KDR (Barleon et al, 1994; Millauer et al, 1993).
Several studies have shown that VEGF genes are induced in the ischemic core and penumbra after middle cerebral artery (MCA) occlusion in rats (Cobbs et al, 1998; Kovacs et al, 1996; Lenmyr et al, 1998). Transient MCA occlusion results in rapid induction of VEGF protein in cortical neurons and pial cells as early as 1 to 3 h (Hayashi et al, 1997, 2003) whereas a slower expression of VEGF in neurons, astrocytes, and macrophages with a peak at day 1 in the ischemic core and at day 7 in the penumbra has been reported after permanent MCA occlusion (Plate et al, 1999).
In parallel with the induction of VEGF, proliferating endothelial cells have been shown to appear as early as 1 day after ischemia, and the number of vessels significantly increases by the 3rd day (Hayashi et al, 2003). Intracerebral infusion of VEGF reportedly stimulates angiogenesis in adult rat cerebral cortex within 3 to 7 days (Krum et al, 2002). Delayed (48 h after ischemia) administration of recombinant human VEGF165 (rhVEGF165) to ischemic rats enhances angiogenesis in the penumbra and significantly improves neurologic recovery (Zhang et al, 2000; Sun et al, 2003). This late recovery-promoting effect of VEGF is thought to be mediated by stimulation of angiogenesis (Zhang and Chopp, 2002).
However, the acute effect of VEGF on cerebral ischemia is more complicated. Zhang et al (2002) found that early postischemic administration (1 h after permanent MCA occlusion) of rhVEGF165 (1 mg/kg intravenously over a 4-h period) to ischemic rats significantly increased blood–brain barrier (BBB) leakage, hemorrhagic transformation, and infarct volume in line with findings showing that VEGF plays a role also in regulating capillary permeability in addition to angiogenesis (Plate, 1999). In contrast, Hayashi et al (1998) found that topical administration of VEGF (9 ng) to the cortical surface reduced infarct size in a 90-min transient ischemia model. Topical application may have unmasked the protective action of VEGF by avoiding its deleterious effect on vascular permeability on intravenous administration. Vascular endothelial growth factor has been shown to have direct trophic effects that can confer neuroprotection independent of angiogenesis (Hayashi et al, 1998; Jin et al, 2000a, b). For instance, VEGF stimulates axonal outgrowth and increases survival of mouse superior cervical and dorsal root ganglion neurons (Sondell et al, 1999), promotes survival of rat mesencephalic neurons in culture (Silverman et al, 1999), and inhibits endothelial cell apoptosis (Alon et al, 1995).
In the present study, we have investigated whether or not intracerebroventricular administration of VEGF soon after reperfusion could replicate the neuroprotective effect observed with topical application, and the mechanism of this protection in a focal ischemia/reperfusion model. We have found that VEGF has a neuroprotective effect possibly mediated by stimulation of neurotrophin receptor-phosphoinositide-3-kinase-Akt pathway with a relatively long (4.5 h after ischemia) therapeutic window. Interestingly, VEGF did not enhance BBB permeability when administered via intracerebroventricular route whereas its intravenous administration enhanced the ischemic injury as previously reported for the rat brain (Zhang et al, 2000).
Materials and methods
Experimental Groups
Swiss albino mice weighing 24 to 33 g were housed under diurnal lighting conditions (12 h darkness and 12 h light) and fasted overnight but allowed free access to water before the experiment. Animal housing, care, and application of experimental procedures were all performed in accordance with institutional guidelines. All efforts were made to minimize animal suffering and to use only the number of animals necessary to produce reliable scientific data. Mice were anesthetized with chloral hydrate (350 mg/kg, intraperitoneally). Body temperature was monitored with a rectal probe and maintained at 37.0+0.2°C by a homoeothermic blanket. Tubing placed in the common carotid artery was used for arterial blood pressure monitoring and heparinization (10 U just before induction of ischemia).
A total of 38 mice were studied. All mice were subjected to 90 mins of proximal MCA occlusion and 24 h of reperfusion. For histological studies, mice were divided into 5 groups (n = 6 for each). Two groups were injected intracerebroventricularly with VEGF164 (Autogenbio***clear, England, 8 ng in 2 μL phosphate-buffered saline (PBS)) 1 or 3 h after reperfusion. Two groups serving as controls were injected only with 2 μL PBS, 1 or 3 h after reperfusion. Vascular endothelial growth factor (15 μg/kg in 0.2 mL PBS, i.e., approximately 0.5 μg per mouse) was intravenously injected to the fifth group 1 h after reperfusion. The dose chosen was reported to increase brain vascular permeability (Tilton et al, 1999) but yet it was much smaller than doses used to promote angiogenesis, hence, giving us the opportunity to elucidate whether or not the reported adverse effect of intravenous VEGF was limited to high doses (Zhang et al, 2000; Manoonkitiwongsa et al, 2004). Eight additional ischemic mice were injected intracerebroventricularly with VEGF or vehicle, and used for Western blotting studies.
Middle Cerebral Artery Occlusion
Proximal occlusion of the right MCA was performed with a nylon filament, as described previously (Huang et al, 1994). Briefly, the right common carotid and external carotid arteries were ligated by a 5–0 silk suture after a midline incision. A nylon filament (8/0) was inserted into the common carotid artery through a small incision proximal to the bifurcation and advanced in the internal carotid artery up to the origin of MCA (10 mm from the bifurcation). The distal 3 mm of 8/0 filament was coated with silicon.
A flexible probe (PF-318 of PeriFlux PF 2B, Perimed Jarfalla, Sweden) was placed over the skull (2 mm posterior, 6 mm lateral to the bregma), away from large pial vessels to monitor the regional cerebral blood flow (rCBF) by laser-Doppler flowmetry. After obtaining a stable 10 mins epoch of preischemic rCBF, the MCA was occluded and rCBF was continuously monitored during ischemia (90 mins) and the first 10 mins of reperfusion. Reperfusion was accomplished by pulling the filament back.
After 24 h of reperfusion, animals were anesthetized with pentobarbital and cardiovascularly perfused with 10% formaldehyde. The brains were carefully removed and sectioned coronally to 2-mm-thick slices starting from the frontal pole. Slices were embedded in paraffin and 5-μm-thick sections prepared from the posterior surface of each slice were processed for Nissl staining and for immunostaining. The infarct area outlined by reduced Nissl staining was measured with image analysis software (NIH Image 1.59). The infarct volume was calculated by multiplying the sum of infarct areas of 5 sequential coronal sections by slice thickness (2 mm).
Intracerebroventricular Injection
Bregma was exposed with a midline incision after fixing the animal's head to a stereotaxic frame. The bone was drilled and removed at 0.9 mm lateral and 0.1 mm posterior to the bregma. A Hamilton needle (25 gauge) was inserted through the burr hole 3.1 mm deep into the brain to reach to the lateral ventricle.
Neurologic Evaluation
Twenty-four hours after recirculation, neurologic deficits were assessed by an observer masked to the identity of treatment and scored as described previously (Huang et al, 1994): 0, no observable neurologic deficits (normal); 1, failure to extend left forepaw on lifting the whole body by the tail (mild); 2, circling to the contralateral side (moderate); 3, leaning to the contralateral side at rest or no spontaneous motor activity (severe).
TUNEL Staining
DNA fragmentation was detected in situ by 3'-OH end labeling using a kit containing terminal deoxynucleotidyl transferase (TdT) and digoxigenin −11-dUTP according to the manufacturer's recommendations (ApopTag; Oncor, Inc., Gaithersburg, MD, USA) Briefly, formalin-fixed and paraffin-embedded 5-μm-thick tissue sections were deparaffinized and dehydrated. Nuclear proteins were stripped from the DNA by incubating in proteinase K for 30 mins, and endogenous peroxidase was blocked with H2O2. Sections were incubated in a buffer containing TdT and digoxigenin-labeled dUTP, followed by digoxigenin-conjugated peroxidase treatment. Diaminobenzidine (DAB) was used as chromagen and the background was stained with methyl green. Positive and negative controls were included. One coronal section passing through the anterior commissure was evaluated for each animal. Brown-colored TUNEL-positive cells with apoptotic nuclear features were counted manually under x 400 magnification in 5 selected brain regions (1, frontal and cingulate; 2, parietal; 3, insular cortices; 4, preoptic area and 5, lateral striatum) (Garcia et al, 1993). Apoptotic neurons were identified by the presence of various types of chromatin condensation or apoptotic bodies. Cells showing diffuse cytoplasmic labeling or light nuclear staining without apoptotic chromatin changes were not counted. The number of TUNEL-positive cells was normalized by dividing the number of TUNEL-positive cells by the infarct area (Elibol et al, 2001; Unal et al, 2001).
Western Blotting for Phospho-Akt
Samples were obtained from the ischemic core of VEGF-treated (3 h after reperfusion) and vehicle groups (n = 4 each). Fresh brain tissues were homogenized in RIPA buffer (20 mmol/L Tris-HCl, pH 7.5, 50 mmol/L NaCl, 1 mmol/L EDTA, 0.5% NP40, 0.5% SDS, 0.5% deoxycholic acid). The homogenate was centrifuged at 14000g for 20 mins at 4°C, and the supernatant was used for analysis. After the same volume of Tris-glycine sodium dodecyl sulfate sample buffer (Invitrogen, Carlsbad, CA, USA) was added to the supernatant, equal amounts of the samples were loaded per lane. Proteins were transferred to PVDF membranes and incubated with anti-phospho-Akt mouse monoclonal antibody (Cell Signaling, Beverly, MA, USA 1:1000). Western blots were performed with horseradish peroxidase-conjugated anti-mouse immunoglobulin G (Chemicon International, Temecula, CA, USA) and were developed using enhanced chemiluminescence kit (Amersham Biosciences, Piscataway, NJ, USA). β-Actin was used as an internal standard. Densitometric measurements are expressed as ratios to β-actin values.
Immunohistochemistry
After 5-μm-thick slices were obtained from paraffin-embedded tissues, they were deparaffinized at 56°C overnight and hydrated in xylol and graded alcohol solutions. The sections were rinsed with PBS (pH 7.4) and nonspecific binding was blocked using 3% normal goat serum (Serotec) in PBS for 20 mins. The sections were then incubated with antialbumin primary antibody (Bethyl Laboratories, Montgomery, Texas, USA, 1:50 in PBS) at 37°C for 120 mins and with biotinylated secondary antibody (Vector Laboratories, Burlingame, CA, USA) for 1 h at room temperature, followed by an ABC process (ABC-Elite kit, Vector Laboratories, Burlingame, CA, USA). Finally, the sections were exposed to DAB (Sigma, St Louis, Missouri, USA) as chromagen. Negative controls were carried by omitting the primary antibody. Immunostained sections were analyzed with a light microscope (Nikon, Eclipse E600). The tissue area showing albumin leakage was measured with image analysis software (NIH Image 1.59). The tissue volume with BBB breakdown was calculated by the same method used in infarct volume calculation.
Statistics
Mean values of arterial blood pressure, rCBF, infarct volume, neurologic disability scores, number of TUNEL-positive cells, tissue volume with BBB breakdown, and densitometric measurements were compared by means of Kruskal–Wallis analysis of variance followed by Mann–Whitney U-test. P-values less than 0.05 were considered to be significant. Mean values in the text are given with their standard deviations.
Results
Arterial blood pressure and rCBF values were not significantly different between the experimental groups during MCA occlusion and reperfusion (Table 1).
Physiologic parameters in experimental groups

Values are given as mean±s.d. Regional cerebral blood flow (rCBF) and mean arterial blood pressure (MABP) values are the average of recordings throughout 90 mins of ischemia and during the first 10 mins of reperfusion.
Infarct Volume and Neurologic Evaluation
Occlusion of the MCA for 90 mins caused an infarct of around 45 mm3 when measured 24 h after reperfusion. Intracerebroventricular VEGF administration 1 h after reperfusion significantly decreased the infarct volume by 35% (29+9 versus 44+2 mm3 in VEGF- and vehicle-treated groups, respectively). Vascular endothelial growth factor administered 3 h after reperfusion was still protective and significantly reduced the infarct size by 46% (27+7 versus 49+7 mm3 in VEGF- and vehicle-treated groups, respectively) (Figure 1A). In contrast to intracerebroventricular administration, intravenously administered VEGF significantly increased the infarct volume to 73+12 mm3 in line with a previous report (Zhang et al, 2000).
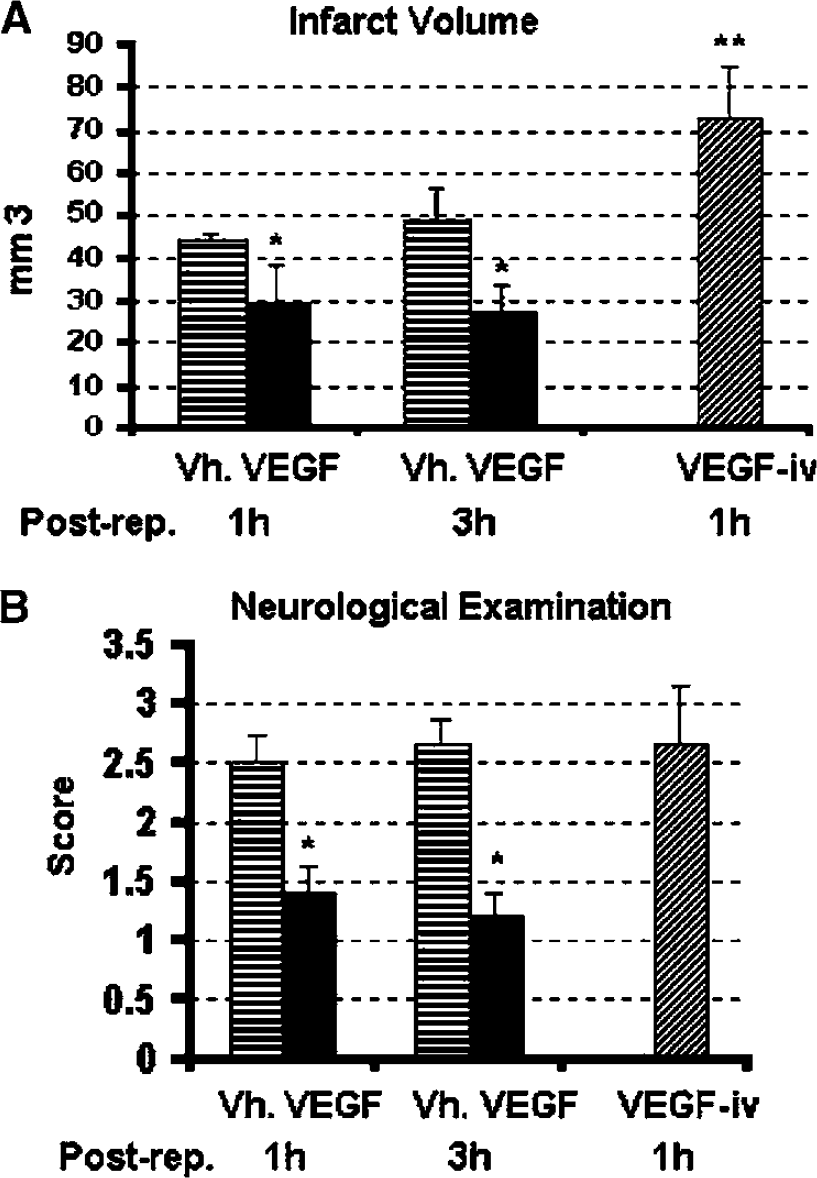
Vascular endothelial growth factor (VEGF) improves stroke outcome when administered intacerebroventricularly but not intravenously. Infarct volume (
Mean neurologic disability scores determined 24 h after reperfusion paralleled the changes in infarct volume in all groups (Figure 1B). Neurologic disability scores were 2.5+0.5 and 2.7+0.5 in vehicle-treated groups injected 1 and 3 h after reperfusion, respectively. Intracerebroventricular administration of VEGF 1 or 3 h of reperfusion significantly decreased the disability scores to 1.4+0.5 and 1.2+0.5, respectively whereas the mean neurologic score of mice treated with intravenous VEGF was 2.7+0.5.
TUNEL-Positive Cells and Phospho-Akt Levels
There was an abundance of TUNEL-positive neurons in the ischemic MCA area on coronal sections passing through the anterior commissure. To eliminate the effect of reduction in infarct size on the number of apoptotic cells, we compared the number of labeled cells per mm2 of infarct area. The number of TUNEL-positive apoptotic neurons was 32+9/mm2 in 3 h vehicle-treated group. Independently to the decrease in infarct size, intracerebroventricularly injected VEGF significantly reduced the number of TUNEL-positive neurons by 52% (to 16+9/mm2) in the 3 h treatment group (P<0.05) (Figure 2). This group also displayed significantly higher Phospho-Akt levels compared with the vehicle-treated group (0.56+1.84 versus 0.27+0.92 densitometric units) (Figure 3).
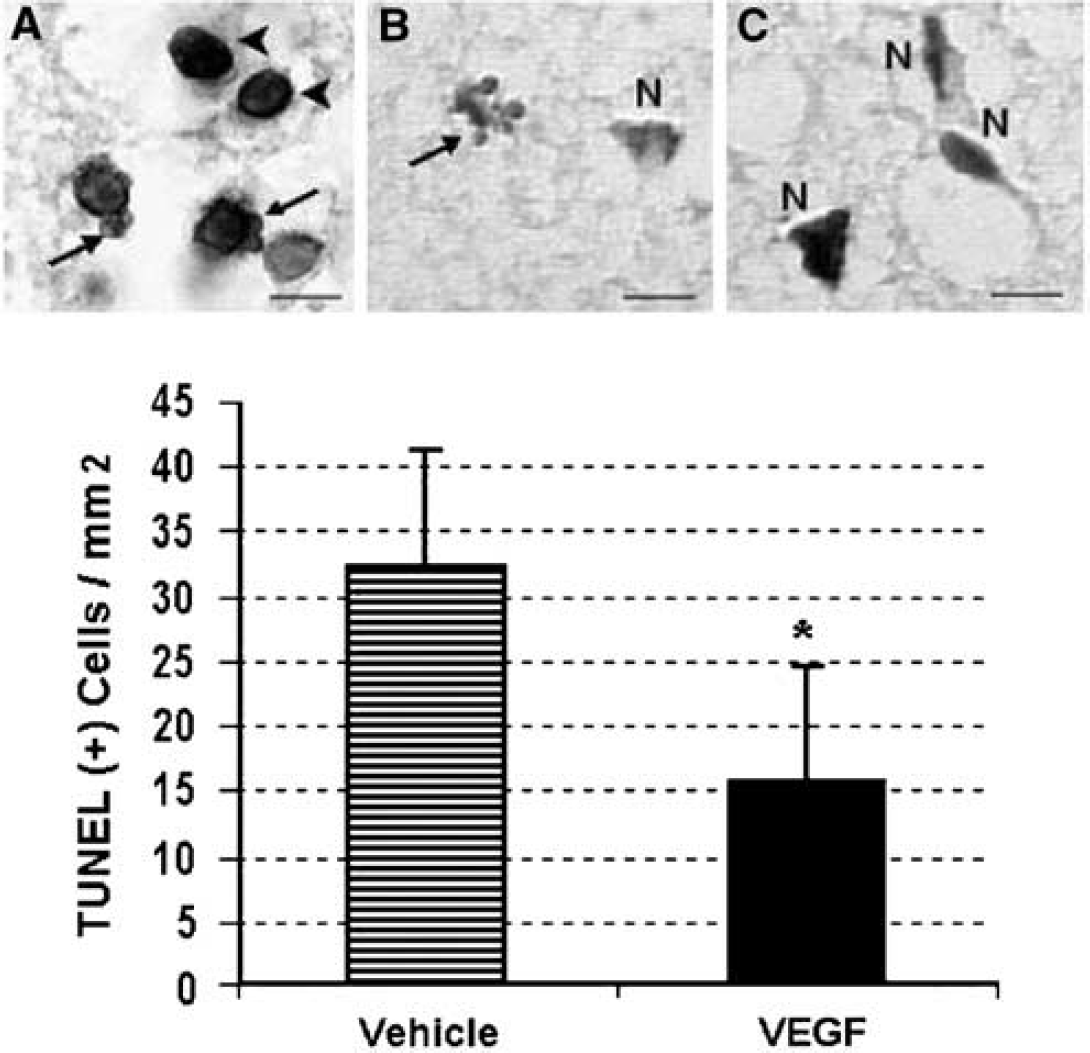
Vascular endothelial growth factor (VEGF) treatment (intracerebroventricularly, 3 h after reperfusion) reduces the number of TUNEL (+) apoptotic cells. Apoptotic cells were identified by TUNEL positivity plus apoptotic features such as peripheral chromatin clumping (arrowheads in
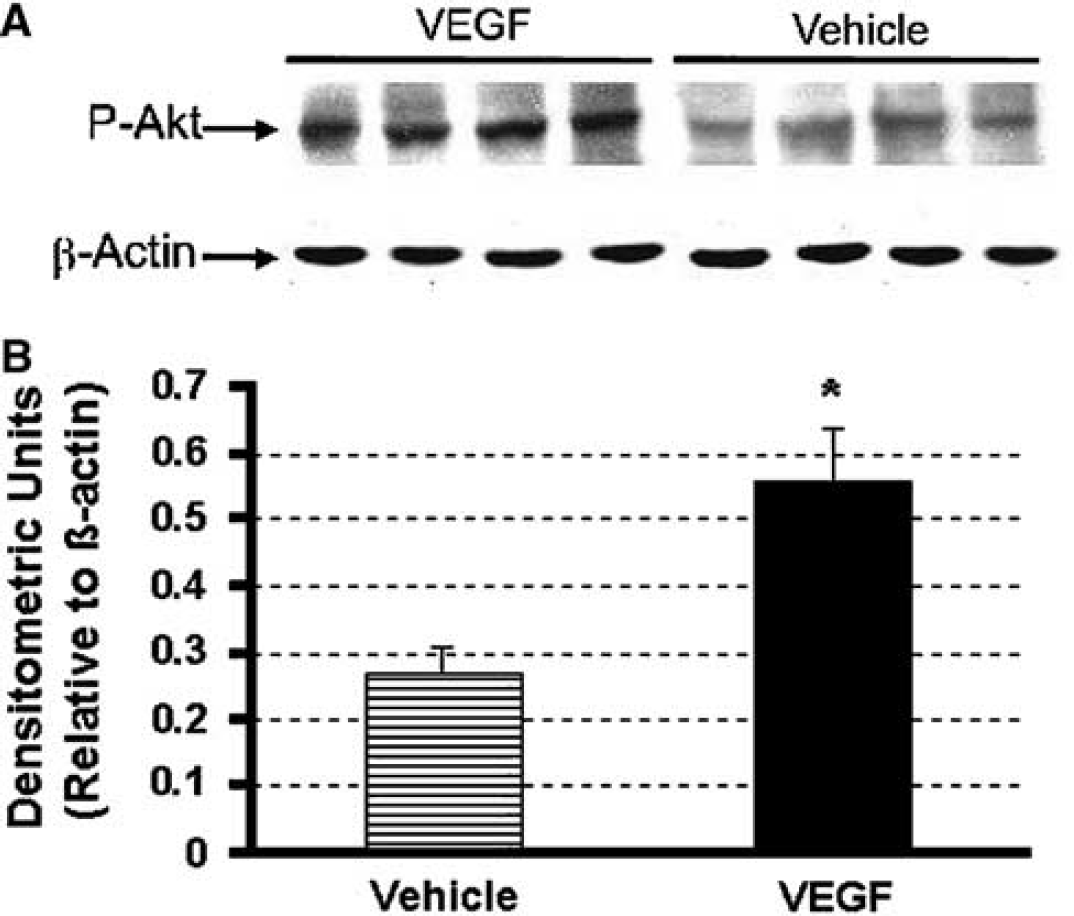
Western blot analysis of phospho-Akt (p-Akt) from ischemic core of vascular endothelial growth factor (VEGF)- and vehicle-treated groups. Vascular endothelial growth factor or vehicle was administered intracerebroventricularly 3 h after reperfusion. Brain samples were collected 24 h after reperfusion. Phospho-Akt levels were higher in VEGF-treated ischemic brains (
Tissue Volume with Blood–Brain Barrier Breakdown
Blood–brain barrier breakdown was assessed by measuring the albumin extravasating ischemic area. The ischemic MCA territory was positively labeled with antialbumin antibody after 90 mins of MCA occlusion and 24 h of reperfusion. No albumin-positive staining was observed in the contralateral hemisphere and in negative control brain sections incubated in the absence of antialbumin antibodies. Brain volume showing albumin leakage was 41+12 and 40+12 mm3 in 1 and 3 h vehicle-treatment groups, respectively. Administration of VEGF intracerebroventricularly 1 or 3 h of reperfusion decreased the tissue volume with albumin leakage to 23+12 and 22+14 mm3, respectively (P<0.05, Figure 4). In contrast, intravenous VEGF injection significantly enhanced the volume of albumin leaking tissue to 64+10 mm3 in parallel with the increase in infract volume (Figure 4).
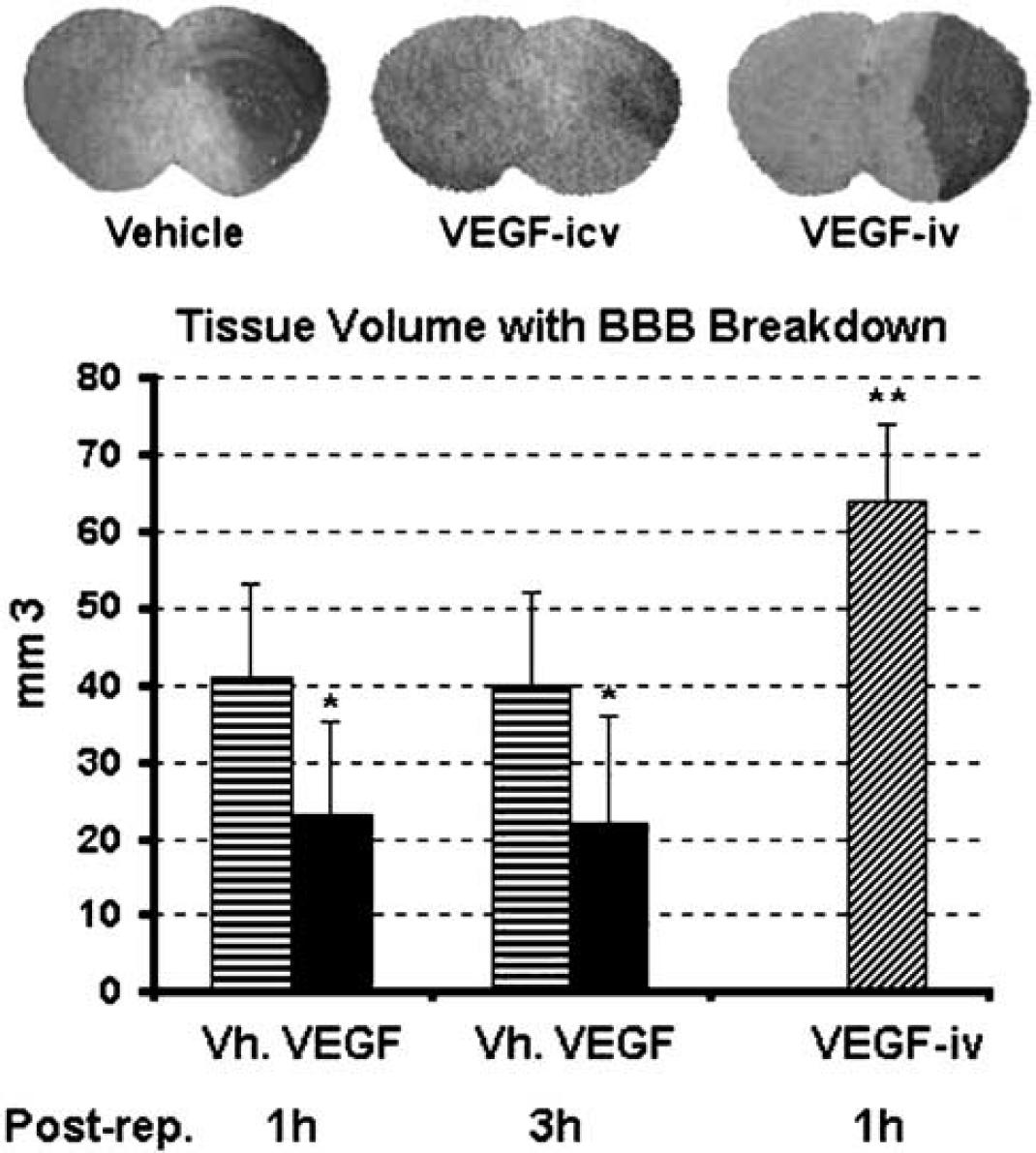
Vascular endothelial growth factor (VEGF) administered intracerebroventricularly decreases ischemia-induced blood–brain barrier (BBB) leakage, contrary to its intravenous administration. Albumin leakage was detected immunohistochemically with antialbumin antibody. Dark stained areas on coronal sections illustrate the middle cerebral artery (MCA) territory with leaky BBB (
Discussion
Early intracerebroventricular administration of VEGF decreased the infarct volume in mice subjected to MCA occlusion. Not only the infarct volume and neurologic scores but also the BBB leakage improved with VEGF treatment. The VEGF-treated animals had higher brain levels of phospho-Akt and less TUNEL-positive apoptotic cells compared with vehicle-treated mice 24 h after ischemia. Taken together, these data suggest that VEGF is able to promote cell survival pathways in the ischemic brain and counteract cell death. Interestingly, this early neuroprotective action had a relatively long therapeutic time window (4.5 h after ischemia, 3 h after reperfusion). In contrast, intravenous administration of VEGF adversely affected the infarct size.
Although delayed administration of VEGF promotes recovery after stroke, our findings suggest an early and direct infarct-sparing effect as well (Sun et al, 2003; Wang et al, 2004; Zhang et al, 2002). In parallel to our findings, VEGF treatment has been shown to provide protection against several forms of neuronal injury independently of its angiogenic action. For example, VEGF exerts neurotrophic effects on cultured adult mouse superior cervical and dorsal root ganglion neurons (Sondell et al, 1999). It also enhances the survival of mesencephalic neurons in organotypic explant cultures (Silverman et al, 1999) and cultured hippocampal neurons subjected to in vitro ischemia (Jin et al, 2000b). Vascular endothelial growth factor protects HN33 cells from death induced by serum withdrawal (Jin et al, 2000a) and reduces hypoxic death of both HN33 cells and cultured cerebral cortical neurons (Jin et al, 2001). Similarly, VEGF protects cultured hippocampal neurons against glutamate (Matsuzaki et al, 2001) and N-methyl-
Treatment with VEGF may protect cells by more than a single means. In one well-established mechanism, VEGF activates phosphoinositide-3-kinase-Akt pathway via neurotrophin receptors. The serine–threonine protein kinase, Akt exerts antiapoptotic effects by preventing the release of cytochrome c from mitochondria, by inactivating fork-head transcription factors (hence, hindering transcription of proapoptotic genes), by inducing transcription of the survival genes regulated by NF-κB and CREB, and by phosphorylating and inactivating the proapoptotic factors BAD and pro-caspase-9 (Chao, 2003). Although we have not examined changes in gene expression in this study, strikingly high levels of phospho-Akt in VEGF-treated ischemic brains supports the idea that Trk receptor-phosphoinositide-3-kinase-Akt pathway was activated as it was reported for neuronal cultures exposed to hypoxia-glucose deprivation (Jin et al, 2000b, 2001).
Exogenous administration of several other neurotrophic factors such as basic fibroblast growth factor (bFGF), neurotrophin 4 (NT 4), or brain-derived neurotrophic factor (BDNF) are also protective in several animal models of cerebral ischemia (Beck et al, 1994; Chan et al, 1996; Kawamata et al, 1996; Schabitz et al, 1997; Tsukahara et al, 1994). Interestingly, Akt is reported to be activated after cerebral ischemia in the rat (Jin et al, 2000c; Friguls et al, 2001) and, endogenous neurotrophic factors such as NT 4 and BDNF are also likely to exert protective effects in cerebral ischemia as mice lacking one allele of BDNF or both alleles of NT 4 are more vulnerable to ischemic injury (Endres et al, 2001). Similarly, rapidly induced VEGF can participate in endogenous early neuroprotective responses in ischemia and may help limiting the infarct size. Recent studies have shown that, like VEGF, other neurotrophins are induced in several pathologies affecting the central or peripheral nervous system (Bartholdi et al, 1997; Lindvall et al, 1992; Tsukahara et al, 1994). However, early neuroprotective or delayed recovery-promoting pathways activated by endogenous neurotrophins seems to be underutilized, allowing room for exogenously administered growth factors to provide further protection.
Ischemia triggers a complex cascade of events leading to cellular swelling and necrosis. However, less severely injured cells die by a process involving caspase activation or by a combination of mechanisms including necrosis and programmed cell death (MacManus and Linnik, 1997). Apoptotic-like DNA fragmentation and TUNEL positivity were demonstrated after focal cerebral ischemia (Li et al, 1995). Although the TUNEL positivity is not highly specific for apoptotic cell death, it suggests activation of the apoptotic cascade when observed together with apoptotic nuclear morphology. Hence, our finding showing a reduced number of TUNEL-positive nuclei displaying apoptotic features in ischemic brains of VEGF-treated animals further strengthens the idea that VEGF, by promoting the pro-survival mechanisms, may counteract apoptotic cell death subroutines activated in ischemic cells. It appears so that promotion of survival factors was merely able to slowdown cell demise in the core region while preventing cell death and saving the tissue in peri-infarct areas. Accordingly, 24 h after ischemia, we observed a smaller infarct volume in VEGF-treated animals along with a reduced number of TUNEL-positive cells in areas destined to infarct.
Unlike neuroprotective agents acting on mechanisms that develops rapidly at the onset of ischemia (e.g., NMDA antagonists), growth factors may have a more favorable treatment window possibly because they act on downstream cell death pathways. Postischemic 4.5 h therapeutic window found in this study for VEGF is significantly longer compared with the therapeutic window of several neuroprotective agents tested in clinical stroke trials so far. Despite this being advantageous, increased BBB leakage observed on intravenous VEGF administration may limit its potential use in humans. However, a single intracerebroventricular injection of VEGF into the normal rodent brain does not induce BBB leakage unlike the peripheral vasculature (Proescholdt et al, 1999). The latter finding is consistent with the neuroprotective effect of VEGF without adversely affecting BBB permeability in our experiments in contrast to its intravenous administration, and suggest that exposure to VEGF from vascular luminal side is required for BBB breakdown. Stimulation of VEGF receptors on the luminal or abluminal side may lead to activation of different signaling pathways. For example, stimulation of luminal receptors may initiate signaling cascades (e.g., nitric oxide might be one of the mediators) that trigger opening of tight junctions or activate matrix metalloproteinases (Tilton et al, 1999; Gu et al, 2002; Zhang and Chopp, 2002). Diffusion kinetics and local concentrations obtained (e.g., intravenously given VEGF may not penetrate brain parenchyma well enough) may also contribute to different outcomes observed with intravenous and intracerebroventricular administrations. Understanding the differences in mechanisms between two routes of administration in future studies may help devise treatment strategies that will overcome the negative impact of VEGF on BBB permeability.
In conclusion, in agreement with studies using topical administration, intracerebroventricular injection of VEGF protects brain against ischemic injury with a relatively long therapeutic time window. This acute infarct-sparing action is likely to be independent of the delayed recovery-promoting effects such as angiogenesis, and possibly involves activation of the PI-3-Akt pathway.
Footnotes
Acknowledgements
The authors are grateful to Dr MA Moskowitz for his support.