
Abstract
Traumatic axonal injury (TAI) is one of the most important pathologies associated with closed head injury, and contributes to ensuing morbidity. The authors evaluated the potential role of calpains in TAI using a new model of optic nerve stretch injury in mice. Male C57BL/6 mice were anesthetized, surgically prepared, and subjected to a 2.0-mm optic nerve stretch injury (n = 34) or sham injury (n = 18). At various intervals up to 2 weeks after injury, optic nerves were examined for neurofilament proteins and calpain-mediated spectrin breakdown products using immunohistochemistry. In addition, fluorescent tracer was injected into the superior colliculi of mice 1 day before they were killed, to investigate the integrity of retrograde axonal transport to the retina. Optic nerve stretch injury resulted in persistent disruption of retrograde axonal transport by day 1, progressive accumulation and dephosphorylation of neurofilament protein in swollen and disconnected axons, and subsequent loss of neurofilament protein in degenerating axons at day 14. Calpains were transiently activated in intact axons in the first minutes to hours after stretch injury. A second stage of calpain-mediated proteolysis was observed at 4 days in axonal swellings, bulbs, and fragments. These data suggest that early calpain activation may contribute to progressive intra-axonal structural damage, whereas delayed calpain activation may be associated with axonal degeneration.
Traumatic axonal injury (TAI) is one of the most common features of closed head injury, suggesting that axons in the white matter are particularly vulnerable to mechanical forces engendered during traumatic insults. The diffuse nature of TAI, however, renders it virtually invisible to conventional imaging techniques in all but the most severe cases of head injury (Mittl et al., 1994). Currently, only postmortem analyses accurately diagnose the extent and nature of axonal injury in humans. To characterize this important aspect of traumatic brain injury, small and large animal models of TAI have been developed. Animal models offer the opportunity to study the evolution of axonal injury, providing unique insight into mechanisms underlying the pathophysiology of TAI. The complexity of brain anatomy and the difficulty of targeting white matter injury to specific tracts, however, have made it difficult to establish causal relations between structural and functional changes resulting from TAI. Furthermore, small animals such as rodents have lissencephalic brains that lack the large proportion of white matter found in human brains. Large animal models, on the other hand, are expensive and labor intensive in analysis.
The optic nerve stretch model of TAI circumvents these obstacles, producing axonal injury in a large, easily accessible population of myelinated CNS axons. This model, first developed in the guinea pig, has been used to demonstrate that TAI results in cytoskeletal disruption, axolemma and myelin damage, interruption in anterograde transport, and transient electrophysiologic alterations (Gennarelli et al., 1989; Tomei et al., 1990; Maxwell et al., 1991, 1995; Maxwell and Graham, 1997; Bain et al., 2001). This ensemble of ultrastructural changes correlates closely with findings reported for axonal injury in humans (Grady et al., 1993; Christman et al., 1994; Sherriff et al., 1994a, b).
In both humans and animals, deformation of the brain during trauma produces a rapid elongation of axons that generally leads not to primary axotomy but to progressive structural damage culminating in secondary axotomy. Disruption of the axonal neurofilament network and impairment of axonal transport with subsequent accumulation of transported proteins and organelles are hallmarks of TAI (Povlishock et al., 1983; Cheng and Povlishock, 1988; Maxwell et al., 1991; Yaghmai and Povlishock, 1992; Pettus and Povlishock, 1996; Povlishock et al., 1997; Maxwell and Graham, 1997). Nonetheless, the intracellular mediators of these events are not well understood. Calpains have been identified as potential initiators of axonal pathology after traumatic injury to the CNS. Recent experiments have demonstrated activation of calpains in axons within minutes after traumatic brain injury (Saatman et al., 1996; Büki et al., 1999b). Some reports, however, suggest that axonal calpain activation is delayed relative to somoden-dritic activation (Newcomb et al., 1997) or occurs only in severely injured axons (i.e., primary axotomy) (Saatman et al., 1996). Therefore, it is unclear whether calpain activation is a necessary step in traumatic secondary axotomy and what role it plays in disruption of axonal transport and cytoskeletal structures. In the present study, we modified the optic nerve stretch injury model for use in the mouse and used this novel model to investigate the temporal relation between calpain activation, neurofilament damage, and impairment of retrograde axonal transport after TAI.
MATERIALS AND METHODS
All procedures involving animals described herein were approved by the University of Pennsylvania Institutional Animal Care and Use Committee and conform to federal guidelines (National Research Council, 1996).
Surgical preparation
Adult male C57BL/6 mice (n = 52; 25 ± 1 g) were anesthetized with 65 mg/kg sodium pentobarbital (intraperitoneally) and placed on a heating pad for surgery. The conjunctiva was separated from the sclera using an incision extending around the entire circumference of the right eye. The extraocular muscles were then cut at their insertion to the eye. The eye was kept moist at all times using sterile saline. A flexible plastic rectangular sling (5 mm x 30 mm) with a longitudinal midline slit was placed underneath the eye with the optic nerve positioned in the slit. The two sides of the slit end were sutured to fix the sling securely behind the eye. The sling was then attached to the optic nerve stretch injury device using a 4–0 suture. Sham-injured mice (n = 18) received anesthesia and the previously mentioned surgical preparation, but did not receive stretch injury.
Optic nerve stretch injury
To receive optic nerve injury, mice (n = 34) were placed in a stereotactic head holder that was positioned on the injury device (modified from Gennarelli et al., 1989) such that the trajectory of the optic nerve at the base of the eye was aligned with the stroke of the solenoid. The sling was connected to a force transducer (ELF T500–1; Entran, Fairfield, NJ, U.S.A.) mounted in series with a solenoid (Lucas-Ledex, Vandalia, OH, U.S.A.) and in parallel with a displacement transducer (Trans-Tek Inc., Ellington, CT, U.S.A.). To ensure consistent loading, a 2.0-g preload, measured by the force transducer, was placed on the nerve. Immediately after obtaining the preload, the solenoid was triggered to produce a rapid, 2.0-mm elongation of the nerve. Force and displacement data were collected as a function of time using custom-made data acquisition software on a personal computer with A/D board (Keithley-Metrabyte, Taunton, MA, U.S.A.). A 2.0-mm elongation was selected to produce a nerve stretch of approximately 20% beyond the initial length of the nerve, a level of injury shown in preliminary studies to consistently produce axonal injury.
Postoperative treatment
For all mice, the sling was removed from the eye and the eyelids were sutured together to prevent drying of the eye. The mice designated for a 24-hour survival were then prepared for injection of Fluorogold (Fluorochrome, Englewood, CO, U.S.A.), a fluorescent tracer. All others were placed on a heating pad until fully recovered from anesthesia, at which time they were returned to their home cages.
Fluorogold injections
To label retinal ganglion cells (RGCs) that were capable of retrograde transport after stretch injury, animals (n = 12 for 1-day survival, n = 11 for 4 days, n = 11 for 14 days) received bilateral injections of Fluorogold into the superior colliculus. On day 0, day 3, or day 13 (24 hours before their designated time of being killed), mice were anesthetized with 65 mg/kg sodium pentobarbital (intraperitoneally) and their heads were shaved. After making a sagittal skin incision to expose the skull, a burr hole was made on either side of lambda over the region of the superior colliculus. Through each burr hole, 2 μL of 2% Fluorogold was injected over several minutes at the level of the superior colliculus. The scalp was sutured and the animal was placed on a heating pad until fully recovered from anesthesia, at which time they were returned to their home cages.
Tissue processing
At either 20 to 30 minutes (n = 2 sham, n = 6 injured), 1 to 2 hours (n = 3 injured), 4 hours (n = 2 sham, n = 5 injured), 1 day (n = 6 sham, n = 6 injured), 4 days (n = 4 sham, n = 7 injured), or 14 days (n = 4 sham, n = 7 injured) after sham injury or stretch injury, mice were anesthetized (65 mg/kg sodium pentobarbital, intraperitoneally) and perfused transcardially with saline containing heparin followed by 10% neutral buffered formalin. The brain was postfixed in situ overnight, after which the optic nerves and eyes were exposed by dissection and separately removed from the head.
Immunohistochemistry on optic nerves
Nerves were cryoprotected in 30% sucrose and then cut longitudinally into 10-μm sections on a sliding microtome (HM400E; Microm, Walldorf, Germany). Free-floating sections were treated with 0.9% H202 in 50% methanol, blocked in 5% normal horse serum with 0.1% Triton X-100, and incubated overnight at 4°C in primary antibody. Sections from each nerve were labeled with the following primary antibodies: anti-NF200 (clone N52, 1:1,000; Sigma Immunochemicals, St. Louis, MO, U.S.A.) that recognizes NF200 regardless of phosphorylation state, SMI32 (1:1,000; Sternberger Monoclonals Inc., Lutherville, MD, U.S.A.) that recognizes dephosphorylated NF160 and NF200, and Ab38 (1:5,000, rabbit polyclonal; a gift from Dr. R. Siman, University of Pennsylvania) that recognizes a spectrin fragment generated specifically by activated calpain (Roberts-Lewis et al., 1994). Primary antibody was detected using biotin-conjugated goat antimouse immunoglobulin G (1:1,000) or goat antirabbit immunoglobulin G (1:2,000; Jackson Immunoresearch, West Grove, PA, U.S.A.) followed by horseradish peroxidase-conjugated streptavidin (Jackson Immunoresearch). The enzymatic reaction was visualized using 3, 3' diaminobenzidine tetrahydrochloride.
Semiquantification of axonal injury
Using a CCD camera (Dage-MTI, Inc., Michigan City, IN, U.S.A.) mounted on a light microscope (Nikon Microphot SA; Optical Apparatus, Ardmore, PA, U.S.A.), sequential, nonoverlapping images were captured along the entire length of one SMI32-immunolabeled optic nerve section per animal. A grid was drawn onto each image, dividing the nerve longitudinally into thirds and subdividing along the length in approximately 100-μm segments. The number of swollen or grossly abnormal axons in each grid element was assessed semiquantitatively using a scale in which 0 = no swellings, 1 = one or two swellings, and 2 = three or more swellings. Ranked scores were assigned to each grid element by each of two separate investigators who were masked to injury status of the animals. Scores from the two investigators were averaged for each grid square. The percent maximal injury was calculated by dividing the sum of all grid element scores by the maximum possible total score (i.e., if all grid elements were ranked as 2). Data were analyzed using a Kruskal-Wallis analysis of variance, followed by individual Mann-Whitney U tests.
Retinal ganglion cell counts
Under a dissecting microscope, a 360-degree incision lateral to the cornea was made in the eye, after which the lens and aqueous humor were removed. The retina was separated from the sclera and four radial cuts were made, spaced at 90 degrees. The retina was then flattened onto a gelatin-coated microscope slide in a small amount of phosphate-buffered saline, and a coverslip was placed. Immediately thereafter, 12 images of each retina were captured under fluorescence microscopy using a DAPI filter set (358 nm EX, 461 nm EM). The 12 regions, each 0.0456 mm2, were imaged at 90-degree intervals at three radial distances from center of the retina, according to previously published protocols (Vorwerk et al., 1996). An investigator blinded to the survival time and injury status of the animal then counted the number of Fluorogold-labeled RGCs in each image for each retina. Counts per field were averaged for each retina and reported as mean number of cells per area for each group. Labeled RGCs were counted for ipsilateral retinae (n = 6 1-day sham, n = 2 14-day sham, n = 5 1-day injured, n = 7 14-day injured) and contralateral retinae (n = 4 1-day injured). Data are presented as means and standard deviations, and were analyzed using a one-way analysis of variance, followed by post hoc Newman-Keuls t-tests.
RESULTS
Profile of axonal neurofilament disruption
Uninjured (sham) nerves showed uniform axonal immunostaining for NF200 (Fig. 1A). Typically, parallel axons traversed a mildly undulated course and were uniform in diameter along their length. Along most of the length of the optic nerve, immunostaining for dephosphorylated neurofilament proteins was very faint (SMI32, Fig. 1B); however, staining increased in intensity near the retinal end of the optic nerve. A relative lack of neurofilament phosphorylation at the proximal end of the mouse optic nerve has been reported previously (Nixon et al., 1994).
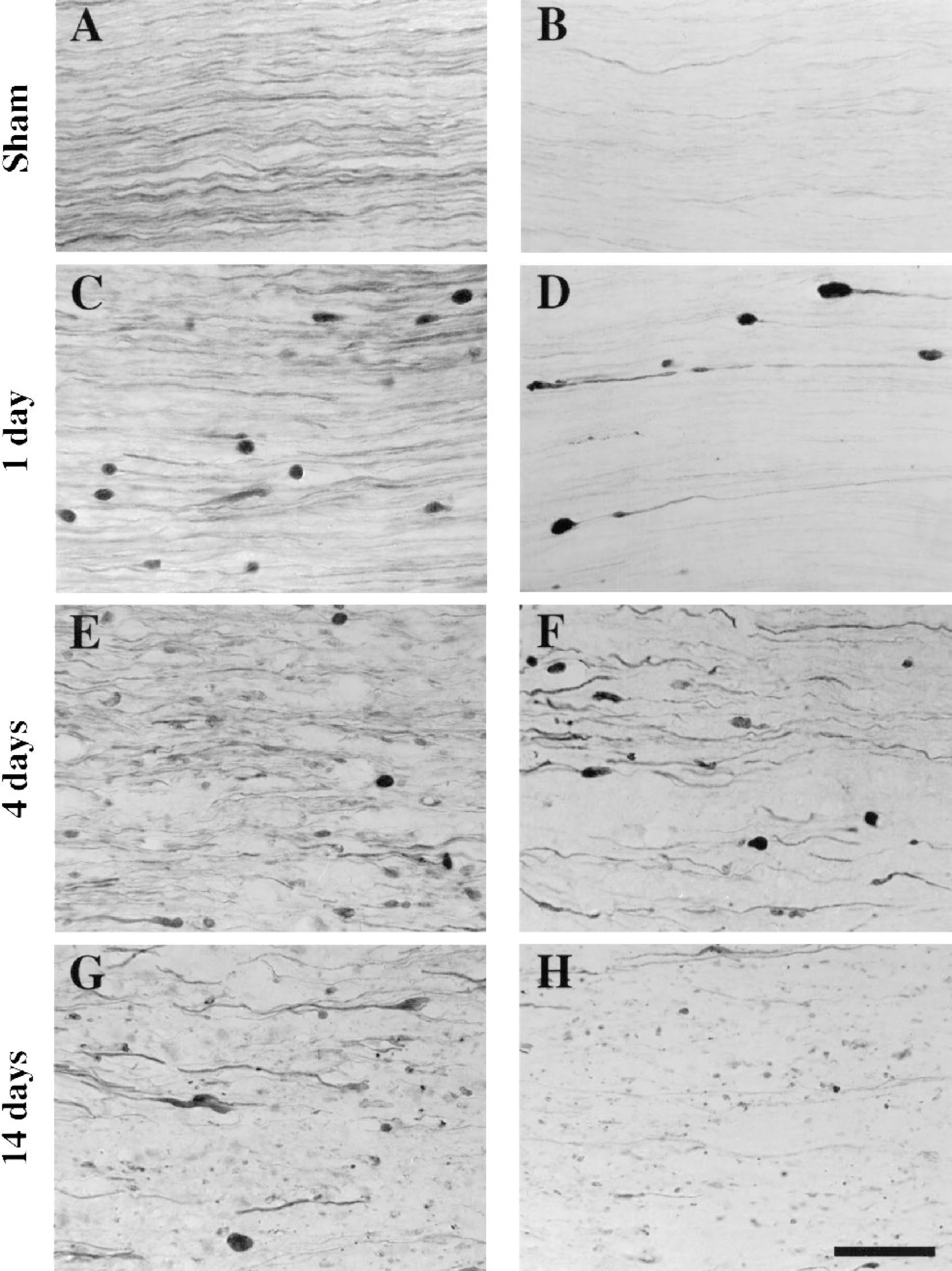
Temporal progression of traumatic axonal injury in the mouse optic nerve. Immunohistochemistry for the heavy neurofilament subunit protein
At 1 day after injury, large axonal swellings and axonal bulbs were observed. Although most axons appeared to have normal morphology and uniform neurofilament (NF) immunostaining, some exhibited intense NF staining within swellings in intact axons and within axonal bulbs indicative of secondary axotomy (Fig. 1C). The axonal swellings and bulbs contained an abundance of dephosphorylated NF (Fig. 1D). The intensity of SMI32 immunolabeling decreased in the axonal segment with increasing distance from the swelling or bulb, consistent with localized dephosphorylation in the injured axon. Axonal bulbs that remained connected to the proximal portion of the axon or to the distal portion of the axon were both observed, suggesting that disconnection may occur on either side of the swelling or bulb.
At 4 days after injury, axons appeared to be disorganized from their parallel orientation and many more axons exhibited NF-immunolabeled swellings than at 1 day after injury. In addition, NF staining was primarily observed in short segments of axons, reflecting early stages of axonal degeneration (Fig. 1E). Dephosphorylated NFs were observed in both axonal swellings/bulbs and in segments of axons with fairly uniform diameter at 4 days (Fig. 1F), suggestive of a more widespread dephosphorylation of NFs than at 1 day after injury.
At 14 days after injury, axonal degeneration and loss of NF immunolabeling were evident (Fig. 1G). Interspersed with large axonal swellings and bulbs were punctate, NF-labeled debris. Much of the NF-containing debris and swellings appeared to contain dephosphorylated NF (Fig. 1H). Although many of the axons within the optic nerve did not exhibit NF disruptions at 1 day after injury (Fig. 1C), the majority of axons within the nerve were clearly damaged at 14 days (Fig. 1G).
Semiquantification of the extent and distribution of axonal injury, using SMI32 immunolabeling as a marker, revealed widespread axonal injury. Axonal swellings and bulbs were found in the central (core) and peripheral regions of the nerve radius and were observed at locations along the entire length of the nerve, with an increased incidence nearer the optic nerve chiasm as compared with the retina (Fig. 2A). Semiquantitative rating of SMI32-labeled axons was effective in discriminating among varying injury severities, as illustrated in Fig. 2A. Furthermore, axonal injury rankings confirmed qualitative observations of axonal damage. The median percentage of axonal damage increased with time, from 14% at 1 day to 65% at 4 days and 86% at 14 days after injury (Fig. 2B). The degree of axonal injury observed at 4 days or 14 days after injury was significantly greater than in sham-injured nerves or in nerves assessed at 1 day after injury (P < 0.05).
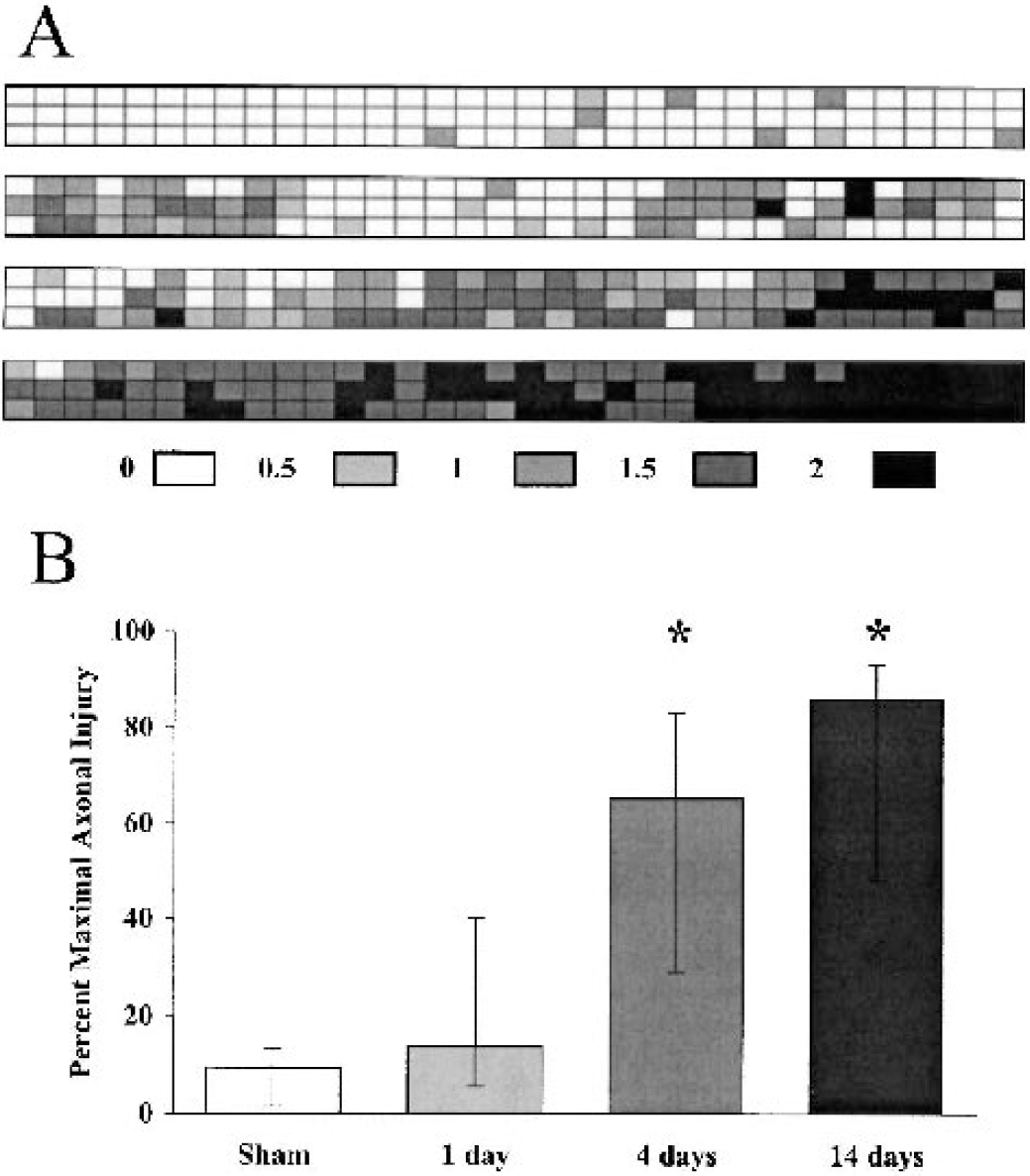
Semiquantitative evaluation of the distribution and severity of traumatic axonal injury in the optic nerve. Grids superimposed on images taken along longitudinal sections of optic nerve illustrate the relative location of injury from the retinal insertion (proximal, left end) to prechiasmatic portion (distal, right end) of optic nerves
Retrograde transport to retinal ganglion cells
In sham-injured animals, Fluorogold injected into the superior colliculus was transported to the retina, resulting in labeling of numerous RGCs (Fig. 3A). At 1 day after optic nerve stretch injury, fewer Fluorogold-positive RGCs were observed in the retina ipsilateral to the injured optic nerve (Fig. 3B). Counts of Fluorogold-labeled RGCs revealed that retrograde transport to the ipsilateral retinae of sham-injured mice (3,497 ± 452) was equivalent to that of contralateral (uninjured) retinae (3,712 ± 402). Furthermore, numbers of RGCs per unit area were consistent with those published previously for mouse retinae (Cenni et al., 1996; Levkovitch-Verbin et al., 2000). A significant decrease, however, in the number of RGCs containing Fluorogold was observed at 1 day after optic nerve stretch injury (1,436 ± 1,084, P < 0.001 relative to sham; Fig. 3C). An equivalent impairment of retrograde transport was observed at 2 weeks after stretch injury (1,547 ± 505, P < 0.001 relative to sham; P = not significant relative to 1 day after injury).
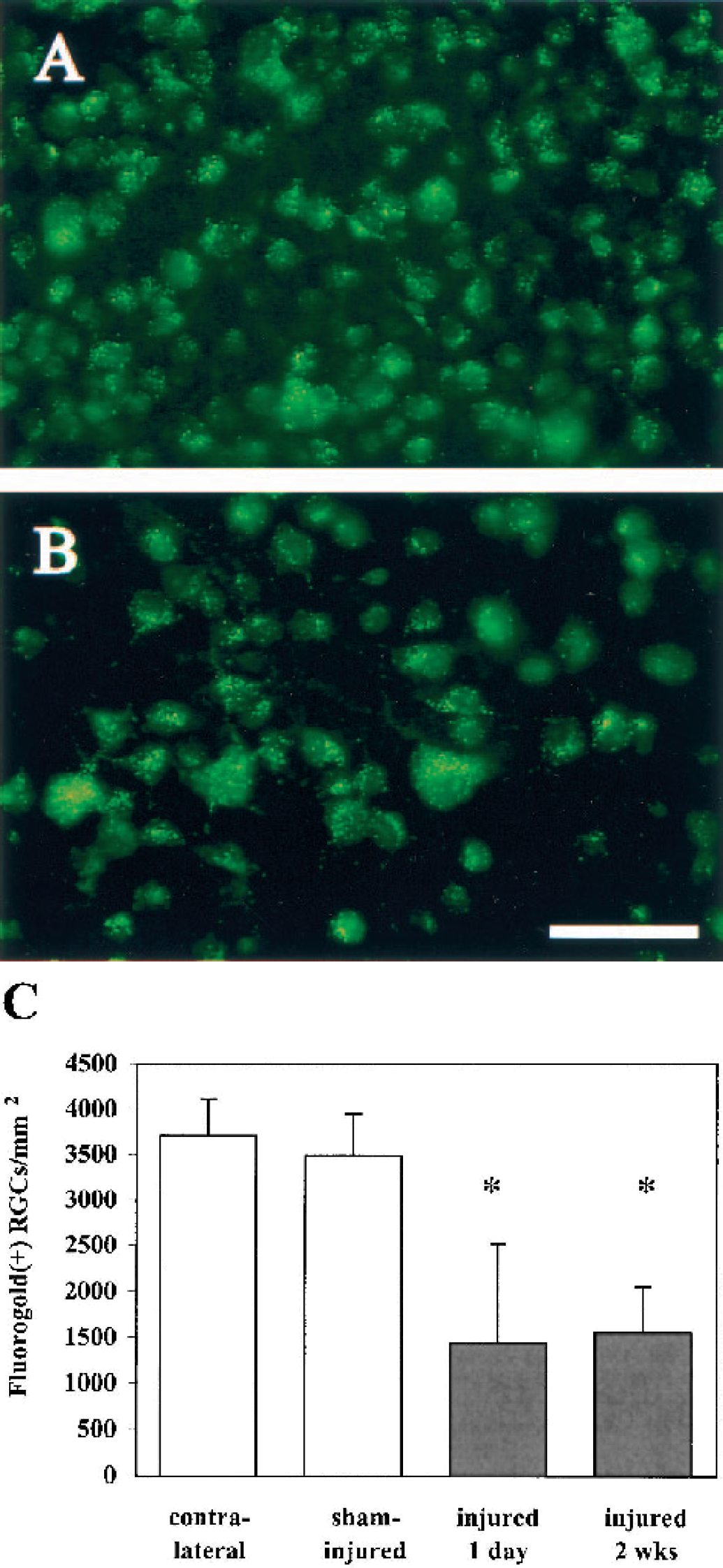
Disruption of retrograde fast axonal transport after optic nerve stretch injury. Compared with whole-mounted retinae from sham-injured controls that exhibited numerous Fluorogold-positive retinal ganglion cells (RGCs) at 1 day after surgery
Calpain-mediated proteolysis of spectrin in injured axons
Using an antibody specific to calpain-mediated proteolytic fragments of spectrin, biphasic activation of calpains was observed after optic nerve stretch injury. After sham injury, no calpain-generated spectrin fragments were observed (Fig. 4). In contrast, in the acute posttraumatic period, numerous intact axons were immunolabeled for calpain-mediated spectrin breakdown products at 20 to 30 minutes (Fig. 4) and 1 to 2 hours (data not shown) after stretch injury. Axons exhibiting proteolytic activity of calpains at these early time points were generally not swollen, but appeared morphologically normal. Furthermore, spectrin breakdown product immunolabeling was observed along the length of the axons, rather than at any focal locations. By 4 hours after injury, axons immunolabeled for calpain-generated spectrin fragments were typically not observed, although two stretch-injured nerves did exhibit limited regions of calpain-specific spectrin proteolysis in intact axons.
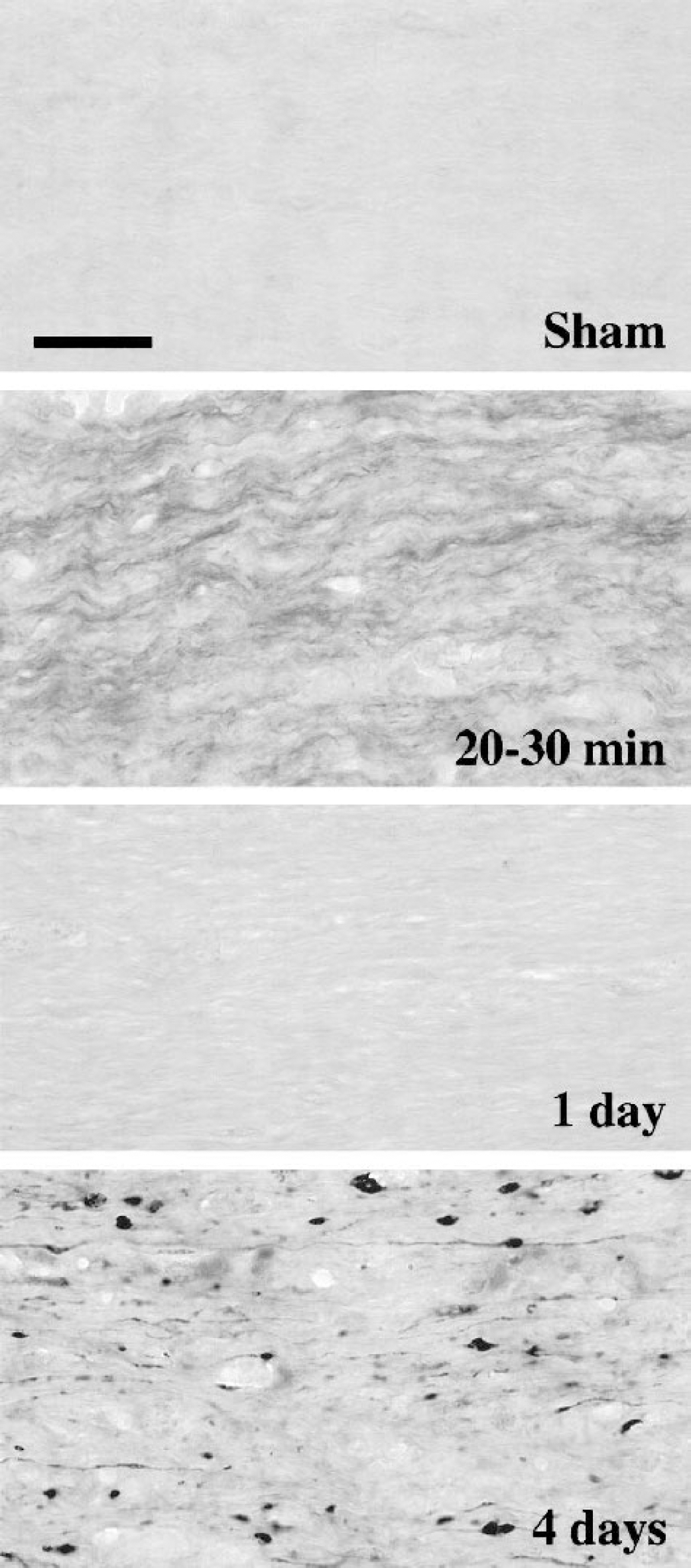
Biphasic calpain-mediated proteolysis of spectrin in traumatically injured axons. Axons in sham-injured nerves showed no evidence of calpain activation. As early as 20 to 30 minutes after stretch injury, however, many undulating, intact axons within injured optic nerves labeled for calpain-generated spectrin proteolytic fragments. Spectrin breakdown by calpain was not evident at 1 day after injury, but was notable in swollen and degenerating axons at 4 days after injury. Scale bar = 25 μm.
Although some neurofilament accumulation and dephosphorylation was observed at 1 day after stretch injury (Figs. 1C and 1D), calpain-generated spectrin breakdown products were not observed at this time point (Fig. 4). A second phase of calpain-mediated spectrin proteolysis, however, was observed at 4 days after injury, characterized by intense immunolabeling of degenerating axons (Fig. 4). Immunostaining for spectrin fragments generated by calpains was localized in numerous small axonal swellings or bulbs and occasionally in thin axonal segments. At 2 weeks after injury, injured nerves exhibited either no spectrin breakdown by calpain or mild immunoreactivity localized to a few scattered spots that appeared to be degenerative debris.
DISCUSSION
We have developed a model of TAI in the optic nerve of mice that applies mechanical deformations relevant to human diffuse axonal injury to a well-defined CNS white matter tract. A rapid, 20% elongation of optic nerve axons induced progressive axonal swelling and neurofilament damage during a 2-week period. Furthermore, persistent impairment of retrograde fast axonal transport was evident by 24 hours after stretch injury. Calpain-mediated proteolysis of spectrin was observed in numerous intact, unswollen axons within 20 minutes to 2 hours after stretch injury, suggesting that calpains may be early mediators of TAI. This initial posttraumatic calpain activity was transient, and was followed by a second phase of calpain activation at several days after injury.
Biomechanical studies using physical models and computer simulations have shown that strains on the order of 15% to 30% may occur in brain regions susceptible to axonal injury after traumatic head injury in vivo (Margulies et al., 1990; Meaney et al., 1995). These estimates of the tissue strain necessary to cause axonal injury have been verified in recent animal experiments (Bain and Meaney, 2000). Therefore, the parameters used for elongation of the mouse optic nerve in the current study maintain biomechanical fidelity with loading conditions shown to produce axonal injury in vivo. Application of a controlled, uniaxial stretch to the optic nerve in vivo resulted in delayed axonal swelling and secondary axotomy throughout the optic nerve. The morphologic features, sporadic distribution, and increased neurofilament immunolabeling of injured axons in the mouse optic nerve closely paralleled axonal pathology described in humans after closed head injury (Grady et al., 1993). Neurofilaments within swollen axons can be detected with either phosphorylation-dependent or independent antibodies (Yaghmai and Povlishock, 1992; Chen et al., 1999). In addition to increased neurofilament immunoreactivity within swellings, however, we also observed neurofilament dephosphorylation in axonal segments that were not overtly swollen, suggesting that disruption of the neurofilament cytoskeleton may occur with or without axonal swelling.
At 1 day after injury, retrograde axonal transport was impaired in as many as half of all optic nerve axons, before evidence of axonal swelling and neurofilament damage in most axons. This suggests that a vulnerable population of axons may exist after TAI that could be prevented from progressing to overt structural damage through therapeutic intervention and that neurofilament disruptions and axonal disconnection may represent the extreme end of the spectrum of TAI. Accumulation of amyloid precursor protein, a protein transported via fast axonal transport, has been demonstrated in axonal swellings as early as 2 to 3 hours after head injury in humans (Sherriff et al., 1994b; Blumbergs et al., 1995; McKenzie et al., 1996), suggesting that transport impairment is an acute posttraumatic event. Accumulation of amyloid precursor protein in damaged axons may precede neurofilament accumulation, or may reflect an aspect of pathology distinct from neurofilament damage (Stone et al., 2000, 2001). Because axonal transport is microtubule dependent, and studies have shown an early loss of microtubules in injured axons (Pettus and Povlishock, 1996; Maxwell and Graham, 1997), it is likely that the early disruption of retrograde transport observed in stretch-injured optic nerves reflects microtubule damage. Interestingly, the number of RGCs with impaired retrograde transport did not change from 1 day to 2 weeks after injury, suggesting that axonal transport dysfunction is an early event that is not progressive. Continuing axonal transport impairment during the first 2 weeks after TAI, however, may also indicate a lack of spontaneous recovery of retrograde transport in injured optic nerve axons.
Increasing evidence supports a role for calpain in the pathology of axonal injury after head injury (Saatman et al., 1996; McCracken et al., 1999; Büki et al., 1999a, b) and spinal cord injury (Li et al., 1995; Schumacher et al., 1999). Wolf et al. (2001) recently demonstrated that dynamic axonal stretch in vitro resulted in increased intra-axonal free calcium concentrations during at least the first 20 minutes after injury. We observed an acute activation of calpains in stretch-injured optic nerve axons within 20 to 30 minutes after injury. Our findings are consistent with previously published studies demonstrating early axonal calpain activation (minutes to hours) in rodent models of traumatic brain injury (Saatman et al., 1996; Büki et al., 1999b). Importantly, we found that TAI induced a biphasic pattern of calpain proteolytic activity, with an early, transient phase in relatively normal-appearing axons followed by a delayed phase in swollen, perhaps degenerating, axons. Biphasic calpain activation has been reported in models of global and focal cerebral ischemia (Saido et al., 1993; Roberts-Lewis et al., 1994; Neumar et al., 2001). Acute, transient calpain activation was induced in, but not restricted to, regions of subsequent cell death. In contrast, delayed, prolonged calpain-mediated proteolysis occurred only in regions of subsequent cell death. These data suggest that transient calpain activation does not necessarily lead to cell death. It is possible, then, that acute, transient calpain activation in stretch-injured axons initiates micro-tubule or neurofilament cytoskeletal modifications or alters signaling pathways, but does not lead irreversibly to axonal degeneration.
The second phase of calpain activation observed at 4 days after injury was concomitant with early signs of loss of neurofilament immunoreactivity and axonal degeneration. By 2 weeks after optic nerve stretch injury, neurofilament-labeled axonal swellings, bulbs, and damaged segments were accompanied by a generalized loss of neurofilament immunoreactivity throughout the nerve, suggestive of neurofilament protein loss accompanying axonal degeneration. Early studies by Schlaepfer et al. linked high calcium concentrations to axonal degeneration and proteolysis of neurofilaments (Schlaepfer, 1974; Schlaepfer and Hasler, 1979; Schlaepfer et al., 1985). More recently, an integral role for calpains in axonal degeneration has been demonstrated (George et al., 1995; Wang et al., 2000). It is likely, then, that the delayed phase of calpain activation observed in stretch-injured optic nerves is associated with proteolysis of axonal neurofilaments. Loss of neurofilament immunoreactivity in regions of axonal injury and calpain activation has been reported in brain-injured rats (Saatman et al., 1996,1998) and in head-injured patients (McCracken et al., 1999).
In conclusion, the mouse optic nerve stretch injury model produces TAI with the morphologic features consistent with clinical cases of diffuse axonal injury. Using this novel injury model, we have demonstrated widespread, acute disruption in retrograde fast axonal transport, followed by disruption of the neurofilament cytoskeleton. Early, transient calpain activation was observed in normal-appearing axons, whereas a second stage of calpain-mediated proteolysis was evident in grossly damaged axons several days after TAI. These findings suggest that calpains may be a critical therapeutic target in preventing axonal damage and dysfunction after trauma, and provide important new information on the potential therapeutic window for inhibition of calpains. Nonetheless, further studies are needed to explore the link between acute calpain activation and the pathology of TAI. The availability of a mouse model of TAI will facilitate the use of transgenic, knockout, and mutant mice for the targeted study of calpains and other candidate mediators of axonal pathology.
Footnotes
Acknowledgements
The authors thank Matthew Leoni and Brent M. Witgen for excellent technical assistance, and Jeanne Marks for assistance in manuscript preparation.