
Abstract
Traumatic brain injury (TBI) is frequently accompanied by a systemic inflammatory response secondary to multiple trauma, shock, or infections. This study investigated the impact of sustained systemic inflammation on cerebral hemodynamics and metabolism in ovine traumatic brain injury. Fifteen sheep were investigated for 14 hours. Head injury was induced with a nonpenetrating stunner in anesthetized, ventilated animals. One group (TBI/Endo, n = 6) subsequently received a continuous endotoxin infusion for 12 hours, whereas a second group (TBI, n = 6) received the carrier. Three instrumented animals served as sham controls. Head impact significantly increased intracranial pressure from 9 ± 4 mm Hg to 21 ± 15 mm Hg (TBI/Endo) and from 10 ± 3 mm Hg to 24 ± 19 mm Hg (TBI) (means ± SD). Internal carotid blood flow increased and cerebral vascular resistance decreased (P < 0.05) during the hyperdynamic inflammatory response between 10 and 14 hours in the TBI/Endo group, whereas these parameters were at baseline level in the TBI group. Intracranial pressure remained unchanged during this period, but increased during hypercapnia. The CMRO2, PaCO2, and arterial hematocrit values were identical among the groups between 10 and 14 hours. It is concluded that chronic endotoxemia in ovine traumatic brain injury was associated with cerebral vasodilation uncoupled from global brain metabolism. Different mechanisms appear to induce cerebral vasodilation in response to inflammation and hypercapnia.
Keywords
Traumatic brain injury (TBI) continues to be one of the leading causes of morbidity and mortality, particularly in younger persons (Ghajar et al., 1995). Cerebral inflammation is nowadays increasingly recognized as a key element in the pathogenesis of secondary tissue damage both in traumatic and ischemic brain injury (Danton and Dietrich, 2003; Morganti-Kossmann et al., 2001). After head trauma, leukocyte infiltration and proinflammatory cytokines are related to disturbances of cerebral blood flow (Siren et al., 2001; Uhl et al., 1994) as well as tissue injury and apoptosis (Fee et al., 2003), and may also contribute to blood-brain-barrier breakdown (Whalen et al., 1998). Less attention, however, has been paid to the cerebral pathophysiology associated with inflammatory processes of primarily systemic origin. Systemic inflammation frequently accompanies brain injury in the context of extensive tissue damage due to multiple trauma (Arand et al., 2001; Muckart and Bhagwanjee, 1997) or secondary to infections (Emsley and Tyrrell, 2002).
In both humans and animals, bacteria or bacterial products such as lipopolysaccharides (endotoxins) trigger the release of proinflammatory cytokines including tumor necrosis factor-α and interleukins 1 and 6. This host response is characterized by a vascular dysregulation that typically leads to profound systemic vasodilation (Hinder et al., 1999; Suffredini et al., 1989). The synthesis of the potent vasodilator nitric oxide by the inducible isoform of nitric oxide synthase (iNOS) plays a key role in this process (Parratt, 1998). In addition, inflammation activates coagulation pathways, and capillary hyperpermeability with subsequent tissue edema develops. It is not known whether or in what way this host response affects the injured brain.
A bolus of endotoxin sufficient to induce short-term systemic vasodilation did not affect the cerebral vasculature in healthy subjects after an observation period of 5 hours (Pollard et al., 1997). However, the way in which the injured brain responds to subsequent inflammation is still not known. Martin and colleagues found that cerebral vascular disturbances after TBI are characterized by a distinct pattern of early hypoperfusion followed by late hyperemia (Martin et al., 1997). This observation resembles those made in the systemic vasculature during systemic inflammation. Whereas hypoperfusion increases the risk for cerebral ischemia, hyperemia has been related to brain swelling, intracranial hypertension, brainstem distortion, and intracranial bleeding (Bouma and Muizelaar, 1992; Martin et al., 1997).
In the present study, it was therefore hypothesized that sustained systemic inflammation induced by continuous administration of endotoxin would lead to cerebral vasoparalysis and aggravation of intracranial hypertension in a modification of a clinically relevant model of TBI in sheep (Lewis et al., 1996).
MATERIALS AND METHODS
With the approval of the Animal Ethics Committee of Münster, Germany, the experiment was performed in 15 adult ewes (44 ± 2 kg).
Animal preparation
The sheep, including three animals that received sham treatment and served as a control group, were instrumented under ketamine anesthesia 24 hours before the experiment. After intramuscular induction of anesthesia, an arterial thermodilution catheter (2015L13, Pulsion, Münich Germany) and a central venous catheter were inserted percutaneously. The common carotid artery was then exposed via a right-sided lateral incision from the thyroid cartilage up to the mandible. After ligation of the external carotid artery, a flow probe (HSB6SB; Transonic, Ithaca, NY, U.S.A.) was attached to the common carotid artery just proximal to the carotid bifurcation. Vessels originating from the internal carotid artery below the cranial base were ligated. The sheep were kept overnight in metabolic cages with free access to water. On the day of the experiment, the animals were continuously anesthetized with S(+)-ketamine (5 mg/kg + 5 mg · kg−1 · h−1) and diazepam (0.2 mg/kg + 0.1 mg · kg−1 · h−1). After additional local anesthesia with 5 mL bupivacaine 0.5%, a tracheostomy tube (ID 8) was inserted. The animals were then paralyzed with 0.2 mg/kg + 0.1 mg · kg−1 · h−1 rocuronium and mechanically ventilated.
After exposure of the skull, two burr holes were made above the right parietal lobe and the sagittal sinus, respectively, with the dura mater left intact. A venous catheter (Insyte 18-G; Beckton Dickinson, Madrid, Spain) was introduced into the sagittal sinus and secured with sutures. In the parietal burr hole, the dura mater was perforated and a bolt introducer kit for intracerebral monitoring probes (IM2; Integra Neurosciences, Andover, U.K.) was inserted. A fiberoptic intracranial pressure (ICP) catheter (Ventrix NL-950, Integra Neurosciences) and a temperature microprobe (Integra Neurosciences) were introduced via the bolt kit. After instrumentation, the animals were allowed to recover for 2 hours but remained under general anesthesia as previously described. Before the head-impact procedure, the intracerebral catheters were removed to avoid mechanical damage, while the sinus catheter remained in place. The parietal burr hole was temporarily closed with bone wax. After head impact, the intracerebral probes were reinserted into the parietal burr hole.
Experimental protocol
The animals were randomly assigned to one of three groups before the experiment was started:
Traumatic brain injury (TBI) (n = 6)
Traumatic brain injury plus endotoxemia (TBI/Endo) (n = 6)
Control group (sham) (n = 3)
The sheep were investigated for 14 hours (Fig. 1). Anesthesia was continued for the entire experimental period as previously described. Mechanical ventilation was adjusted to maintain normoxia and normocapnia by variation of the respiration rate and the FiO2.
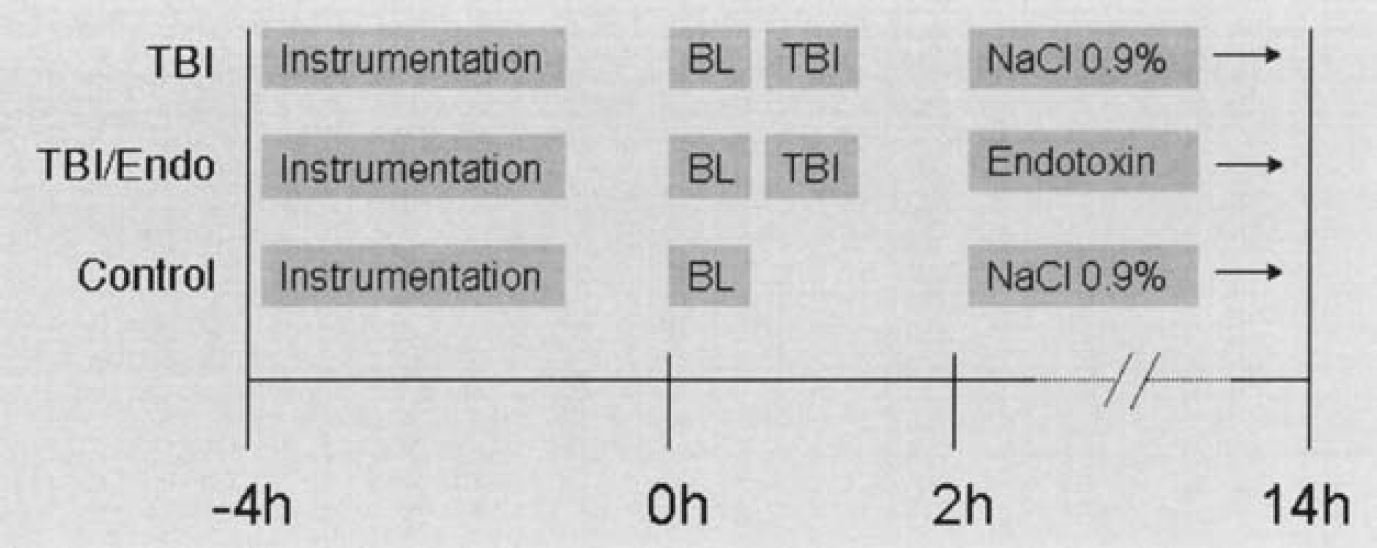
Experimental protocol. Instrumentation: tracheostomy and insertion of intracerebral catheters (after a recovery period of 2 hours); BL, baseline; TBI, traumatic brain injury.
After baseline measurements (0 hours), head injury was induced. The sheep were placed in prone position with the head resting on a support to allow free lateral movements of the head during the impact. To prevent skull fractures, a steel plate (6-mm thick) that was individually cut out to exactly fit between the supraorbital process and the external auditory meatus, was attached to the left temporal fossa of the sheep skull. In contrast to the method described by Lewis et al. (1996), the steel plate was placed above the shaved skin and tightly fixed with elastic cords that were pulled through small burr holes in the plate. This modification was made to avoid an additional surgical procedure during the experiment. A left-temporal head impact was then delivered by a mechanical stunning device (MK 1200; Schermer, Ettlingen, Germany), which is approved for euthanasia of domestic livestock. A captive bolt with a mushroomshaped head is propelled from the muzzle of the stunner against the skull by the discharge of blank cartridge inserted in a chamber behind the proximal end of the bolt. The velocity of the head of the stunner depends on the charge selected. In contrast to the previous study, a higher charge was used to deliver the impact (No. 17 instead of No. 13). The animals in the control group did not undergo a head-impact procedure.
Measurements were performed after a recovery period of 1 hour (2 hours) after head injury. An infusion of Ringer's lactate was adjusted to maintain the individual central venous pressure (CVP) measured at 2 hours for the next 12 hours. After the measurements at 2 hours, the animals in the TBI/Endo group were continuously infused with endotoxin (lipopolysaccharide from Salmonella typhi, 10 ng · kg−1 · min−1) while the animals in the TBI and control groups received the carrier (NaCl 0.9%). Blood glucose and potassium levels were kept within normal limits by substitution, when necessary. PaCO2 was tightly controlled by end-expiratory CO2 monitoring and blood gas analysis. At the end of the experiment after 14 hours, the animals were deeply anesthetized with 50 mL of intravenous propofol 1% and killed by injection of 30 mL saturated KCl solution. The brain was immediately removed and fixed in formalin for later neuropathologic examination.
Data collection
For the experiment, the catheters were connected to pressure transducers (Ohmeda Ltd, Erlangen, Germany) and continuously flushed with a heparin-saline solution (5 IU · mL−1). Zero calibration of the arterial catheters was performed at the level of the hip joint of the front leg, with the animals supine. The internal carotid blood flow (ICBF) was measured continuously with a Doppler flowmeter (HT-311, Transonic). ICP and brain temperature values were obtained using bedside monitors (Ventrix NL 950–100 and Licox CMP, Integra Neurosciences). The cardiac index and systemic vascular resistance indexes were calculated using standard formulas. The body surface area was derived from the body weight. The static pressure autoregulation index (sARI) was tested by slow continuous infusion of norepinephrine to induce an increase in mean arterial pressure by 20 mm Hg at 0, 2, and 14 hours, and was calculated as shown below. At the end of the experiment, the cerebral reaction to hypercapnia was tested by increasing arterial CO2 to 50 mm Hg over 10 minutes.
The cerebral hemodynamic and metabolic indexes were calculated as relative values using the ICBF (in liters per minute) instead of CBF:
CPP = MAP – ICP
CVR = CPP/ICBF
CMRO2 = (arterial oxygen content – sinus oxygen content) × ICBF
CMRLac = (sinus lactate concentration – arterial lactate concentration) × ICBF
sARI = (%ΔCVR/% ΔABP)
where CPP is cerebral perfusion pressure; MAP is mean arterial pressure; ICP is intracranial pressure; CVR is cerebrovascular resistance; ICBF is internal carotid blood flow; and sARI is the static autoregulation index.
Electrolyte concentrations, arterial hematocrit, and arterial and sinus blood gas analyses were performed using a blood gas analyzer (ABL 520; Radiometer, Willich, Germany)
Tissue processing
After autopsy, the brains were fixed in 10% buffered formalin for 14 days. Six cerebral regions were excised from each brain (left temporal, right parietal, left parietal, left frontal, left occipital, and brainstem), routinely processed, and paraffin embedded for further histologic assessment. Tissue sections 10–μm thick were stained with hematoxylin-eosin. Immunohistochemistry was performed on 2–μm sections for neuron-specific enolase (NSE monoclonal; Dako, Hamburg, Germany) at a dilution of 1:200. NSE immunostaining labels injured axons after brain injury as early as 1.5 hours after impact (Ogata and Tsuganezawa, 1999) and morphologically highlights axonal retraction balls/swollen axons as a morphological correlate of traumatic axonal injury. The avidin–biotin complex method with diaminobenzidine as a chromogen was used to detect the reaction product.
Histologic examination
Within the six tissue blocks selected from each brain, axonal retraction balls/swollen axons in the areas of interest positively stained by NSE antibody were qualitatively assessed by a consultant neuropathologist masked to the study groups. Additional morphologic findings such as intracerebral hemorrhage, subarachnoid hemorrhage, and brain edema were also noted.
Statistical analysis
Nonparametric statistical tests were used throughout due to the small number of animals in each group. Mann-Whitney tests were calculated for differences between the TBI and the TBI/Endo groups. Friedmann tests followed by post hoc Wilcoxon tests were applied to evaluate significant effects within these two groups. In contrast to the TBI and TBI/ENDO group, head injury was not performed in the sham (control) group. The absence of brain injury is likely to have influenced the level of sedation in the controls compared to the treatment groups. Major differences in sedation, however, preclude direct comparisons in terms of cerebral blood flow and metabolism. The data from the control group were therefore not included into the statistical analysis. Data are expressed as means and standard deviations. Statistical significance was defined as P < 0.05.
RESULTS
Neuropathology
Traumatic brain injury was characterized by a similar pattern of intracranial pathologies in both treatment groups (Table 1). In the control group, small lesions were confined to the parietal lobe of the right hemisphere, where intracerebral probes were placed via the introducer bolt. This type of finding was considered as artificial in all groups. Subarachnoid hemorrhage in the treatment groups occurred in all brain regions, with the extent of bleeding tending to be more pronounced at the side contralateral to the impact (contrecoup). Small intracerebral hemorrhages followed this pattern of distribution. Diffuse brain edema was visible microscopically in all head-injured sheep. Traumatic axonal injuries were distributed inhomogeneously among the sheep brains. Although this pathology was virtually absent in the left hemispheres, marked axonal injuries were seen in the parietal and temporal lobes of the right hemisphere and, in an even more pronounced way, in the brainstem (mainly in the dorsolateral quadrants, the corticospinal tracts, the medial lemnisci and the central tegmental tracts). Contusions were not a typical feature in this model of TBI. In one animal in the TBI group, however, bilateral frontoparietal contusions occurred and were associated with particularly high intracranial pressure. No skull fractures were identified during the autopsies.
Neuropathologic findings
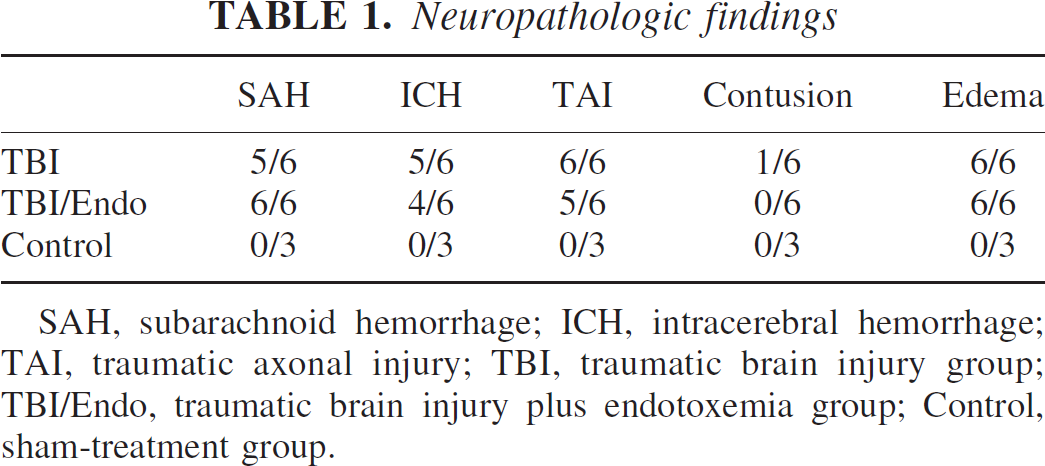
SAH, subarachnoid hemorrhage; ICH, intracerebral hemorrhage; TAI, traumatic axonal injury; TBI, traumatic brain injury group; TBI/Endo, traumatic brain injury plus endotoxemia group; Control, sham-treatment group.
Cerebral circulation and metabolism
The index of cerebral pressure autoregulation (sARI) varied in the normal range between 0.7 and 1 before and after head injury. Global static cerebral autoregulation was therefore assumed to have remained intact in this model of traumatic brain injury. In response to the head impact, the ICBF (Fig. 2) initially decreased significantly in both treatment groups, and this was not accompanied by a concomitant change in cerebrovascular resistance (CVR) (Fig. 2). Endotoxemia further decreased ICBF at 3 hours in the TBI/Endo group (P < 0.05 3 hours vs. 0 hours). Although the ICBF then returned to baseline level in the TBI group, it increased markedly in the presence of a significant decrease in CVR in the later phase of endotoxemia between 10 and 14 hours in the TBI/Endo group (P < 0.05 vs. 0 hours and vs. TBI). Cerebral venous oxygen saturation (SCVO2) (Fig. 2) increased during this condition in TBI/Endo (P < 0.05 vs. TBI).
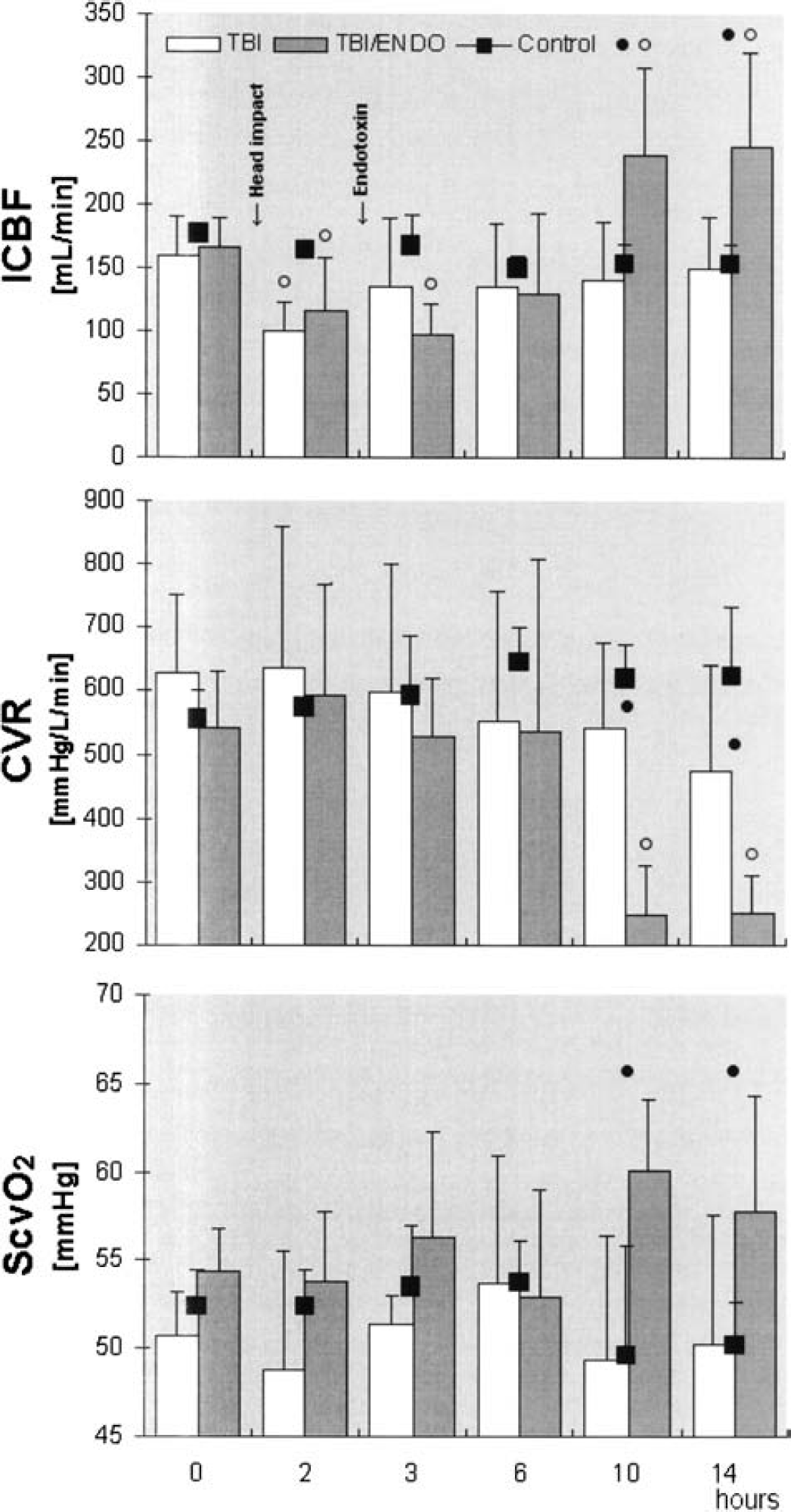
ICBF, internal carotid blood flow; CVR, cerebral vascular resistance; SCVO2, cerebral venous oxygen saturation; TBI, traumatic brain injury group; TBI/Endo, traumatic brain injury plus endotoxemia group. Open circle = P < 0.05 vs. 0 hours (Wilcoxon); closed circle = P < 0.05 TBI vs. TBI/ENDO (Mann-Whitney).
Increased blood flows in TBI/Endo were not associated with an increase in the CMRO2 between 10 and 14 hours (Fig. 3). After 3 hours, however, CMRO2 was significantly lower during endotoxemia (P < 0.05 vs. TBI). Cerebral lactate production (CMRLac) significantly increased during endotoxemia at 10 hours (P < 0.05 vs. TBI) but returned to the level of the TBI group after 14 hours. Arterial blood glucose concentrations and intracerebral temperatures are shown in Table 2.
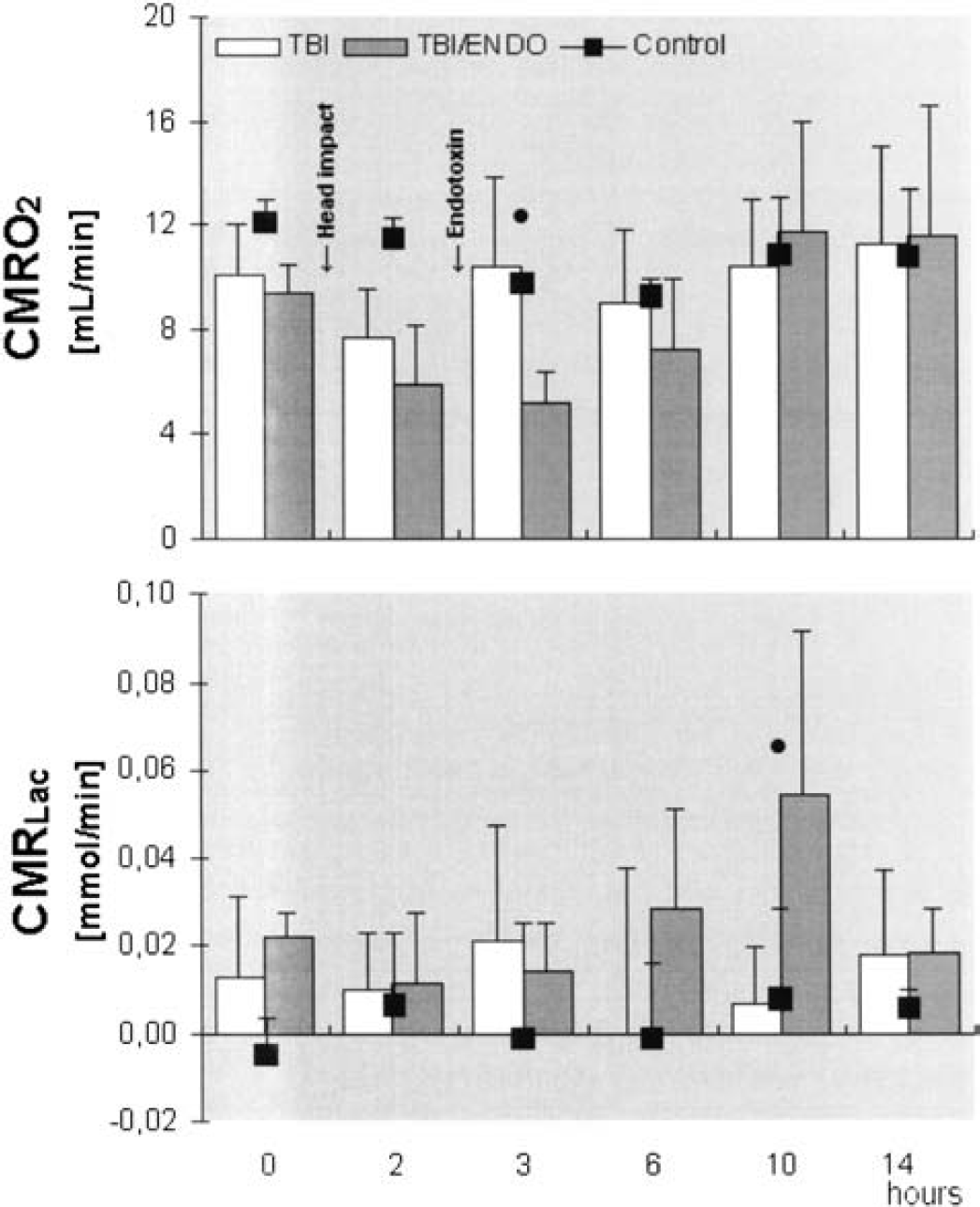
CMRLac, cerebral metabolic rate of lactate (cerebral lactate production); TBI, traumatic brain injury group; TBI/Endo, traumatic brain injury plus endotoxemia group. Closed circle = P < 0.05 TBI vs. TBI/Endo (Mann-Whitney).
The head impact resulted in a marked increase in ICP in the TBI group (from 9 ± 4 to 21 ± 15 mm Hg) and in the TBI/Endo group (from 10 ± 3 to 24 ± 19 mm Hg) after 2 hours (P < 0.5 vs. 0 hours) (Fig. 4). ICP remained significantly elevated for the rest of the observation period, without significant differences between the groups. In the control group, ICP was stable at normal levels throughout the experiment. Because of the increase in ICP, cerebral perfusion pressure declined significantly in both treatment groups (Fig. 4). As endotoxemia was associated with decreasing mean arterial pressures (MAPs), cerebral perfusion pressure tended to be lower in the TBI/Endo group than in the TBI group. At 3 and 10 hours, this effect reached statistical significance. Hypercapnia increased ICBF and ICP in all groups (Fig. 5).
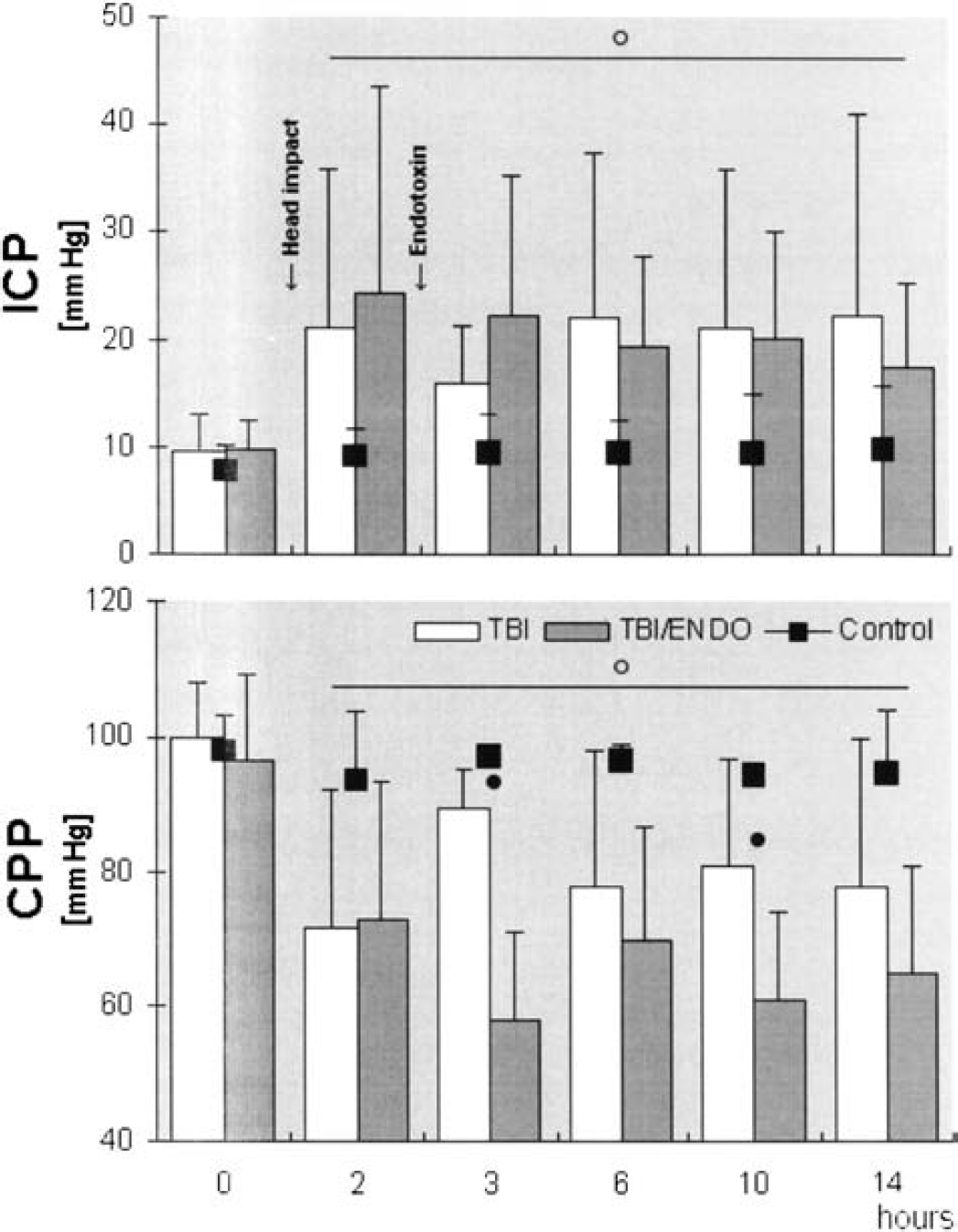
ICP, intracranial pressure; CPP, cerebral perfusion pressure; TBI, traumatic brain injury group; TBI/Endo, traumatic brain injury plus endotoxemia group. Open circle = P < 0.05 vs. 0 hours (Wilcoxon); closed circle = P < 0.05 TBI vs. TBI/Endo (Mann-Whitney).
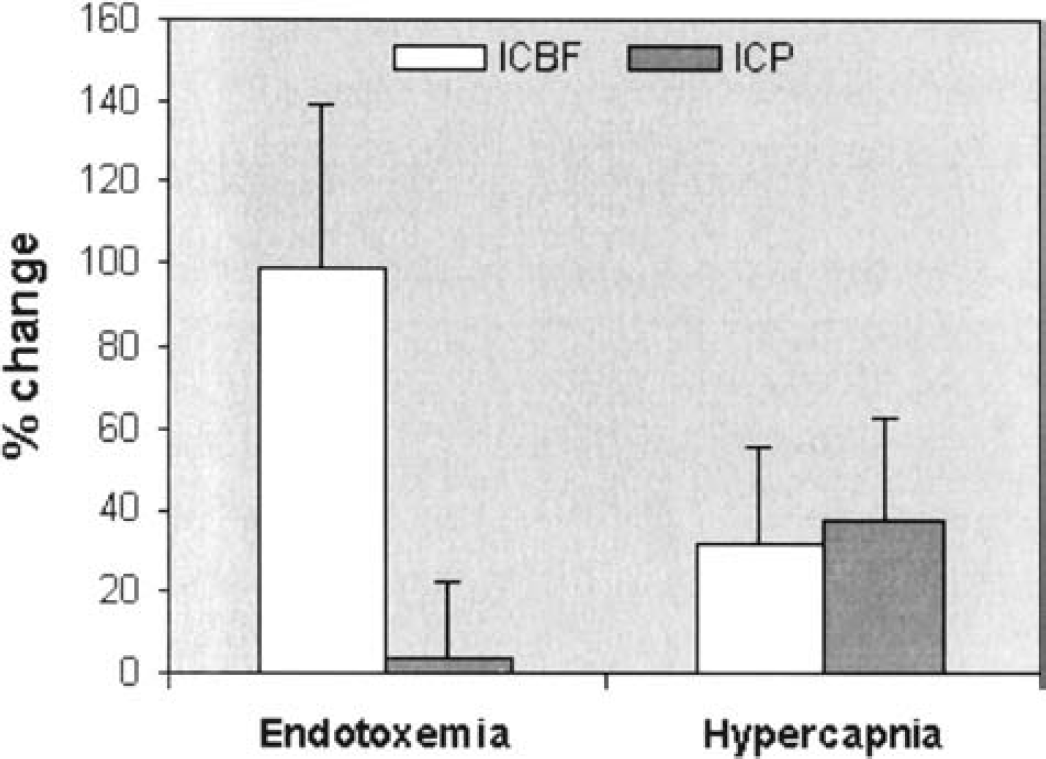
Percentage changes in the internal carotid blood flow (ICBF) in comparison with changes in intracranial pressure (ICP) during endotoxemia (8 hours vs. 12 hours) and hypercapnia (40 mm Hg vs. 50 mm Hg).
Systemic circulation and gas exchange
The head impact was not accompanied by major changes in systemic hemodynamic parameters, except for a significant decrease in MAP in the TBI group at 2 hours (Table 3). In the TBI/Endo group, the systemic circulation showed a hyperdynamic response between 8 and 12 hours of endotoxemia (Table 3). This was characterized by an increase in the cardiac index and a decrease in the systemic vascular resistance index (SVRI) (both P < 0.05 vs. 0 h and vs. TBI). The MAP reached a significantly lower level in comparison with baseline and with the TBI group in this period. In the control group, systemic hemodynamic variables were stable throughout the observation period. Gas exchange parameters were kept at physiological levels in all groups (Table 3).
DISCUSSION
The main findings in this study were that head impact in sheep induced traumatic axonal injury, intracranial hemorrhage, and sustained intracranial hypertension. Subsequent endotoxemia was associated with a systemic inflammatory response that markedly increased internal carotid blood flow in the presence of a profound decrease in cerebrovascular resistance. In contrast, global cerebral oxygen metabolism and intracranial pressure were unaffected by endotoxemia in this model.
Rationale for this model
In the present study, a modified sheep model of traumatic brain injury introduced by Lewis and colleagues was used (Lewis et al., 1996). Its characteristics include the simultaneous occurrence of traumatic axonal injury and intracranial hemorrhage, which represent typical pathologies associated with human traumatic brain injury (Hardman and Manoukian 2002). In contrast to the findings of Lewis and colleagues, the ICP remained increased throughout the observation period of 14 hours, while ICP had returned to baseline level after 6 hours in their study. This discrepancy probably resulted from the higher charge used to deliver the impact in the present study. Increased ICP developed secondary to intracranial hemorrhage and cerebral edema. There was considerable variation in ICP after the head impact within the groups, as indicated by high standard deviations. However, neither the pattern of cerebral pathologies nor the hemodynamic effects of endotoxemia largely differed among animals with different degrees of intracranial hypertension.
In larger animals such as sheep, it is possible to use standard brain monitoring. In addition, continuous infusion of low-dose endotoxin in sheep is sufficient to induce a hyperdynamic cardiovascular pattern characterized by profound vasodilation, elevated cardiac output, and hypotension (Traber et al., 1988). In this investigation, a hyperdynamic systemic inflammatory response is described for the first time in anesthetized and mechanically ventilated animals with traumatic brain injury. Anesthesia was maintained by continuous intravenous ketamine in combination with diazepam in order to maintain cardiovascular stability. On the other hand, ketamine potentially influences glutamate-dependent neurotoxicity via blockade of NMDA-receptors (Allen et al., 1999) and may alter CBF and ICP. We cannot generally exclude such effects, as non-anesthetized brain injured or endotoxemic animals have not been investigated in this study. In the Control group, however, there were no changes in ICP or CBF after induction of ketamine anesthesia during mechanical ventilation. This finding is in keeping with data from experimental and clinical studies that suggest that increases in CBF and ICP can be avoided by mechanical ventilation (Pfenninger et al., 1985) and additional administration of benzodiazepines as a co-anesthetic (Bourgoin et al., 2003).
Intracerebral temperature and blood glucose
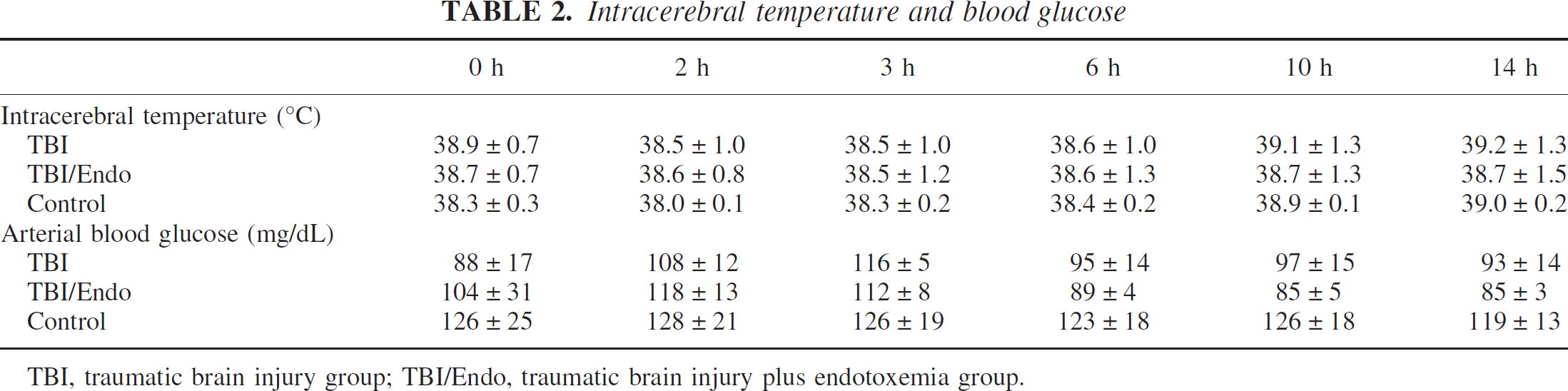
TBI, traumatic brain injury group; TBI/Endo, traumatic brain injury plus endotoxemia group.
Cerebral circulation and metabolism
A marked decline in cerebrovascular resistance (CVR) accompanied the decrease in the SVRI after 8 to 12 hours of systemic inflammation in the TBI/Endo group. The decrease in the SVRI has been previously described during endotoxemia and can be attributed to the effects of inflammatory mediators (Dal Nogare, 1991; Groeneveld et al., 1997). In the unaffected brain, metabolic coupling and pressure autoregulation tightly adjust cerebral vascular tonicity to metabolic requirements. The major determinants of CBF including CMRO2, arterial CO2, and hematocrit were identical among the groups between 10 and 14 hours. These data are therefore consistent with cerebral vasodilation being uncoupled from the global metabolism in response to systemic inflammation. Although the decrease of CVR in the TBI/Endo group may have partly resulted from declining MAP in the presence of intact pressure autoregulation, lower MAP does not explain the simultaneous increase in the ICBF.
Previous studies focusing on the cerebral effects of a bolus of endotoxin have provided divergent results (Moller et al., 2002; Pollard et al., 1997). Firstly, in contrast to the present study, healthy subjects were investigated. It appears possible that the presence of brain injury may have influenced the cerebral effects of endotoxin. Traumatic brain injury triggers local inflammatory cascades (Ghirnikar et al., 1998) and may thereby render the brain more vulnerable to a subsequent endotoxemic insult (“double hit”) (Siren et al., 2001). In sheep, our group has previously shown that the potentiation of systemic inflammation by a preceding insult leads to organ dysfunction and death (Hinder et al., 2003). Secondly, the endotoxin was continuously infused over a prolonged period. The cerebral effects of endotoxin may vary depending on the dosages used and the periods studied. The release of nitric oxide by iNOS is likely to play a key role in inflammatory vasodilation (Parratt 1998). In the rat brain, among other tissues, iNOS/mRNA induction peaks at 6 hours after an endotoxin challenge (Wong et al., 1996). This observation correlates well with the massive decrease in CVR after 8 hours of endotoxemia in the present study.
Thirdly, concerns may be raised regarding the use of ICBF as a measure for unilateral CBF. Blood flow in the internal carotid artery can partly be directed to extracerebral tissues via the ophthalmic artery or intracavernous branches, and redistribution of flow within the circle of Willis via communicating arteries may occur (Soustiel et al., 2003). The lack of bilateral measurements of internal carotid artery flow or direct measurements of global CBF is a potential weakness in the present study. Despite these possible sources of inherent error, the unilateral ICBF correlates excellently with global CBF measured by microsphere deposition and radionuclide techniques (Meadow et al., 1999; Schoning et al., 1994). Moreover, the significant increase in the SCVO2 in the TBI/Endo in comparison with the TBI group at 10 and 14 hours is in keeping with cerebral hyperperfusion during endotoxemia.
Hemodynamics and gas exchange
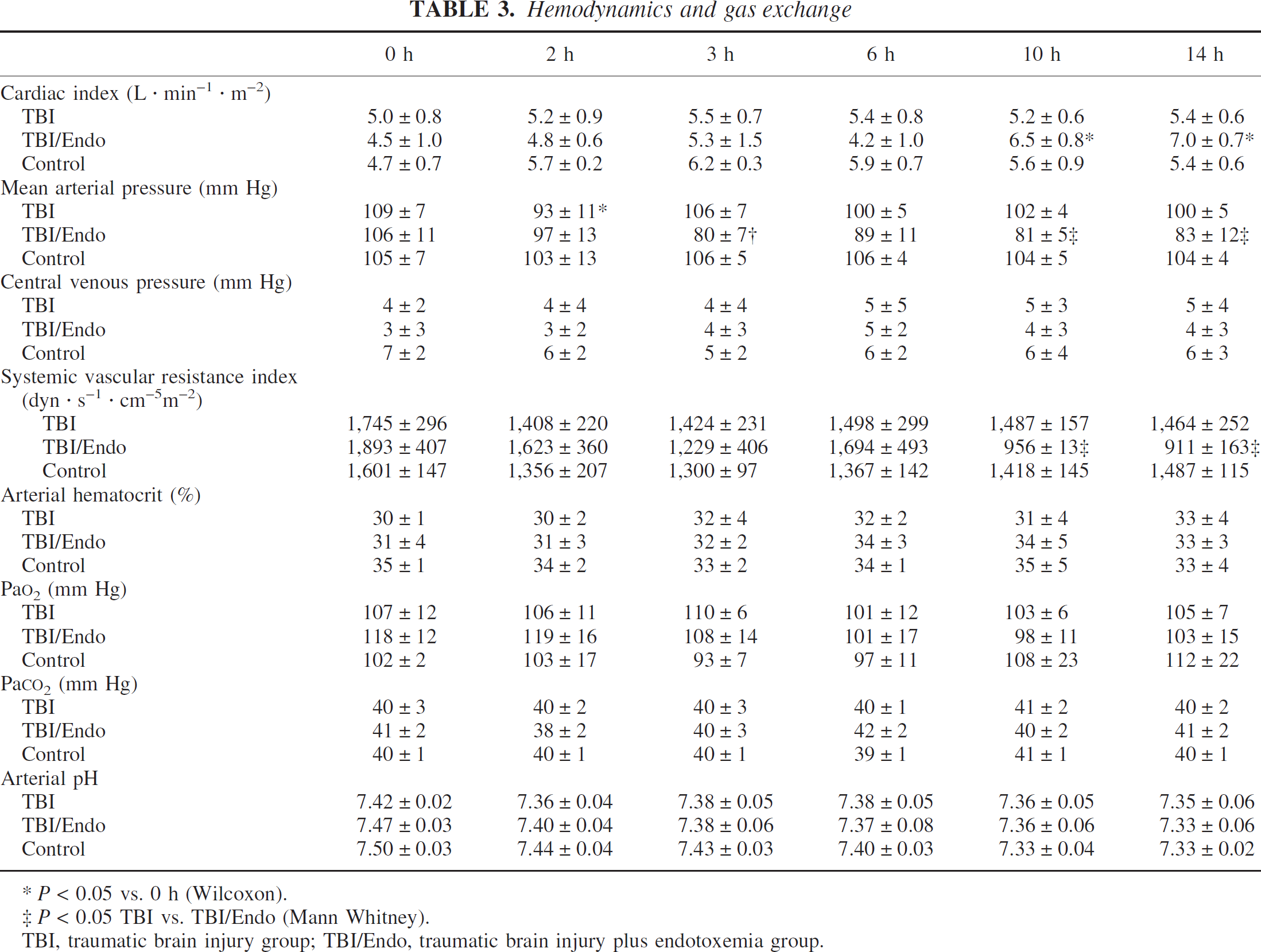
P < 0.05 vs. 0 h (Wilcoxon).
P < 0.05 TBI vs. TBI/Endo (Mann Whitney).
TBI, traumatic brain injury group; TBI/Endo, traumatic brain injury plus endotoxemia group.
Surprisingly, the decrease in the CVR apparently had no influence on the development of ICP, despite an approximate 60% increase in ICBF. This type of situation might occur when intracranial compliance is still sufficient to compensate for changes in cerebral blood volume (CBV). ICP, however, was already elevated in response to brain injury. This indicates that intracranial compliance was already exhausted before endotoxemia further increased the CBF. Even in animals that showed particularly high ICP values, intracranial hypertension was not aggravated by hyperperfusion. In contrast, increases in ICBF induced by hypercapnia led to an immediate increase in ICP. Several mechanisms may account for this phenomenon. Bouma and Muizelaar (1992) showed that dissociation between CBF and CBV occurs, which would be consistent with a stable ICP despite elevated ICBF. Although hypercapnia almost exclusively dilates small arterial resistance vessels located within the brain, inflammation might preferably dilate larger vessels adjacent to the brain tissue. The CBV might then differ due to the vessel surface area and the localization of the dilated vessel. The rapidity of changes in CBF may have also played a major role. The slower increase in ICBF in response to endotoxemia may have allowed compensatory changes via the ventricular system. Moreover, the possibility cannot be excluded that the increase in ICBF partly resulted from a passive phenomenon secondary to increased cardiac output. In accordance with Poiseuilles law, the CBV might have remained constant if vascular tonicity remained constant and cerebral intravascular pressures increased. In head-injured patients, however, there is no such correlation between cardiac output and CBF, regardless of the status of cerebral pressure autoregulation (Bouma and Muizelaar 1990).
The present study did not focus on microcirculatory changes and tissue oxygenation after brain injury and inflammation. During sepsis, organ perfusion decreases despite maintained global blood flows (Sielenkamper et al., 2001)—a phenomenon that is most likely attributable to coagulopathy and arteriovenous shunting. In the TBI/Endo group, cerebral lactate production significantly increased despite elevated ICBF at 10 hours. Whether this finding indicates tissue hypoxia during systemic inflammation is unclear, because lactate production returned to the level of the TBI group after 14 hours.
Future studies will need to address possible mechanisms of secondary brain injury at the microcirculatory level, as well as the role of the local host response during systemic inflammation, which may be potentially destructive for the injured brain (Iadecola, 1997; Morganti-Kossmann et al., 2001).
CONCLUSION
Sustained systemic inflammation induces cerebral vasodilation uncoupled from pressure autoregulation and global oxygen metabolism in this clinically relevant model of TBI. The distinct reaction of ICP to increases in CBF may be due to vasodilation at different sites of the cerebral vasculature or may depend on the rapidity of changes in CBF. Further investigations are needed to assess whether inflammatory cerebrovascular dysregulation may be involved in the pathogenesis of secondary injury after head trauma.
Footnotes
Acknowledgements
The authors would like to thank Dr. Adrian W. Gelb (San Francisco, CA, U.S.A.) for the critical review of this manuscript and Dr. Bianca Dräger (Münster, Germany) for statistical support.