
Abstract
Vascular endothelial growth factor-B (VegfB) is an angiogenic protein related to VegfA, although it acts on a different set of tyrosine kinase receptors. Like VegfA, VegfB is expressed in the brain and is induced at sites of brain injury. VegfA has neuroprotective and angiogenic effects, but VegfA-knockout mice die in utero, so the effect of endogenous VegfA signaling in neuropathologic states, such as cerebral ischemia, cannot be tested directly. In contrast, VegfB-knockout mice survive to adulthood with little abnormality in the absence of pathologic stresses. To determine if VegfB regulates the severity of cerebral ischemia, the middle cerebral artery was occluded in VegfB-knockout, heterozygous, and wild-type mice, and the volume of the resulting cerebral infarcts and associated impairment of neurologic function were measured. Infarct volume was increased by approximately 40% and neurologic impairment was more severe in VegfB-knockout mice, implying that endogenous VegfB acts to protect the brain from ischemic injury. VegfB also protected cultured cerebral cortical neurons from hypoxic injury, suggesting that its protective action is mediated at least in part through a direct effect on neurons.
Vascular endothelial growth factor (Vegf, or VegfA) is an angiogenic peptide that enhances endothelial-cell proliferation during development and in response to hypoxia or ischemia. VegfA is also found in neurons (Jin et al., 2000a; Yang et al., 2002) and astrocytes (Krum and Rosenstein, 1998; Salhia et al., 2000), where its expression is increased after focal cerebral ischemia in the rat (Gu et al., 2001; Jin et al., 2000a; Marti et al., 2000) and in patients after stroke (Issa et al., 1999; Slevin et al., 2000). Intracerebroventricular VegfA decreases infarct volume and brain edema after transient focal ischemia (Harrigan et al., 2002), whereas VegfA antisense oligodeoxynucleotides reduce VegfA expression and increase infarct volume (Yang et al., 2002). VegfA protects cortical neurons and HN33 (mouse hippocampal neuron x neuroblastoma) cells from hypoxia and glucose deprivation (Jin et al., 2000c) and HN33 cells from serum withdrawal (Jin et al., 2000b) in vitro. These findings indicate that VegfA has neurotrophic and neuroprotective as well as angiogenic properties; recent studies also point to a role for VegfA in stimulating neurogenesis (Jin et al., 2002; Sun et al., 2003b). Most effects of VegfA are mediated through the Vegfr2/Flk1 receptor tyrosine kinase. Elucidating the roles of other Vegf family members and receptors in cerebral ischemia may help identify new therapeutic targets in stroke.
VegfB, a heparin-binding growth factor with strong homology to VegfA and lesser homology to placenta growth factor (Plgf) (Grimmond et al., 1996; Olofsson et al., 1996), is expressed most abundantly in heart, skeletal muscle, pancreas and brain (Lagercrantz et al., 1998; Li et al., 2001; Olofsson et al., 1996). Its distribution differs from that of VegfA (Nag et al., 2002; Olofsson et al., 1996), although coexpression is observed in some tissues, and VegfB can form both homodimers and VegfA/VegfB heterodimers (Olofsson et al., 1996). The alternatively spliced VegfB167 and VegfB186 isoforms bind to at least two Vegf receptors: Vegfr1/Flt1 (Olofsson et al., 1998) and neuropilin-1 (Nrp1) (Makinen et al., 1999). VegfB promotes angiogenesis after surgically induced hindlimb ischemia in mice (Silvestre et al., 2003) and may play a role tumor angiogenesis because, like VegfA, it is induced in tumors (Gunningham et al., 2001a; Gunningham et al., 2001b; Salven et al., 1998). Mice with targeted deletion of VegfB have defects in cardiac development and function (Aase et al., 2001; Bellomo et al., 2000) and in hypoxia-induced pulmonary vascular remodeling (Wanstall et al., 2002). However, the physiologic role of VegfB in brain remains unknown.
We used VegfB-knockout mice to investigate whether VegfB plays a role in ischemic brain injury. Outcome was assessed by measuring brain infarct volume and neurologic function after middle cerebral artery occlusion (MCAO). Our results indicate that VegfB-knockout mice exhibit an increase in the severity of ischemic brain injury, consistent with a neuroprotective effect of endogenous VegfB.
MATERIALS AND METHODS
VegfB-knockout mice
Experiments were conducted according to policies of the National Institutes of Health. The VegfB gene was disrupted in AB2.1 embryonic stem cells (derived from 129S7/SvEvBrd-Hprtb-m2) and selected cells were injected into C57BL/6-Tyrc-Brd/c-Brd blastocysts (Bellomo et al., 2000). VegfB-knockout mice were obtained from Jackson Laboratories (Bar Harbor, ME, U.S.A.) and bred in-house to produce homozygous knockout (VegfB−/−), heterozygous (VegfB−/+), and wild-type (VegfB+/+) mice. Mice were genotyped by PCR of tail-tip genomic DNA using the allele-specific polymerase chain reaction (PCR) primer 1, 5′-TTCAGGGAGTCTTGGCACTC-3′ (forward primer, which lies above the insertion site); PCR primer 2, 5′ -CTCTGTGTAGCCCTGGCTGT-3′ (reverse common primer, which lies downstream of the insertion site); and PCR primer 3, 5′-AAATGGCGTTACTTAAGCTAGCTTGC-3′ (primer LTR2, which lies at the end of the LTR of the insertion sequence). The PCR protocol included a 3-minute preincubation at 94°C, followed by 35 cycles of 30 seconds at 94°C, 1 minute at 68°C, and 1 minute at 72°C.
Western blots
Protein was extracted from mouse brain as described elsewhere (Jin et al., 2002); 100 μg per lane was electrophoresed by 12% SDS-PAGE and transferred to polyvinyldifluoridine membranes. Membranes were blocked with 5% nonfat dried milk in phosphate-buffered saline (PBS) and 0.1% Tween 20 and incubated overnight with goat polyclonal anti-VegfB (R&D, Minneapolis, MN, U.S.A.; 1:100). Blots were washed with PBS/0.1% Tween-20 and incubated at room temperature for 60 minutes with horseradish peroxidase-conjugated donkey-anti-goat secondary antibody (Santa Cruz Biotechnology; 1:2,000). Peroxidase activity was visualized with a chemoluminescence substrate (NEN, Boston, MA, U.S.A.).
Immunohistochemistry
Mice were perfused with 0.9% saline followed by 4% paraformaldehyde in PBS (pH 7.4). Paraffin-embedded, 6-μm sections were deparaffinized with xylene and rehydrated with ethanol. Double-label immunohistochemistry was performed as described elsewhere (Sun et al., 2001). The primary antibodies were goat polyclonal anti-VegfB (R&D; 1:100), mouse monoclonal anti–neuron-specific nuclear protein (NeuN) (Chemicon, Temecula, CA, U.S.A.; 1:500), mouse monoclonal anti-CD146 (Chemicon; 1:100), mouse monoclonal anti-α2 (vascular smooth muscle) actin (Maine Biotechnology, Portland, ME, U.S.A.; 1:100), and mouse monoclonal anti–glial fibrillary acidic protein (Sigma, St. Louis, MO, U.S.A.: 1:500). The secondary antibodies were rhodamine-conjugated rat-absorbed donkey-anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA, U.S.A.; 1:200) and fluorescein isothiocyanate (FITC)-conjugated anti–goat IgG (Jackson ImmunoResearch; 1:200). Controls included omitting the primary or secondary antibody. Fluorescence signals were detected by Nikon PCM-2000 laser-scanning confocal microscopy.
RT-PCR
Total RNA from mouse forebrains (n = 3 per condition) was prepared using RNeasy Minikits (Qiagen, Valencia, CA, U.S.A.). DNA-free total RNA (2 μg per reaction) was reverse-transcribed into first-strand cDNA using the Reverse Transcription System and Oligo-dT12–18 (Invitrogen, Carlsbad, CA, U.S.A.) as described previously (Clark et al., 2000). The primer sequences, VegfB forward primer 5′-ACCAGAAGAAAGTGGTGCCATG-3′ (284–305) and VegfB reverse primer 5′-TGAGGATCTGCATTCGGACTTG-3′ (499–478), were designed from the mouse VegfB sequence (Genbank accession NM011697). Mouse α-actin sense and antisense primers were used as controls for determining the quantity of RNA. PCR products were separated on 3% agarose gels using 100-bp ladder DNA standards as a size reference.
Vascular morphology, morphometry, and blood–brain barrier function
After anesthesia with 10% chloral hydrate (80 mg/kg), mice were perfused with 100 mL of 10-U/mL heparin in saline and 100 mL of 4% paraformaldehyde in PBS. Carbon black, suspended in an equal volume of 20% gelatin in H2O, was injected into the ascending aorta. The brain was removed and fixed in 4% paraformaldehyde in PBS for 24 hours. Cerebral vasculature was examined under a dissecting microscope (Murakami et al., 1998). To quantify vessel density, 100x images from frontal cortex were acquired using a Nikon Eclipse-800 microscope, DXM1200 digital camera and software (Sun et al., 2003b). Carbon black–stained areas were determined by image analysis using Simple PCI software (Compix, Philadelphia, PA, U.S.A.); areas of staining were identified in the control sample and used to define representative ranges of values for black pixels. Fields from frontal cerebral cortex (three or four fields per animal, three animals per condition) were collected and the area covered by pixels of the defined intensities was calculated, averaged across animals, and expressed as mean percent vessel area per 100 μm2. The integrity of the blood–brain barrier was assessed by immunohistochemistry with biotinylated goat anti–mouse IgG (Vectastain Elite ABC, Vector; 1:200).
Ischemia
Permanent MCAO was induced by intraluminal occlusion with a nylon monofilament suture (Longa et al., 1989). Mice weighing 25 to 30 g were anesthetized with 2.0% isoflurane in 30% O2 and 70% N2O using a vaporizer. Rectal temperature was maintained at 37° ± 0.5°C with a thermostat-controlled heating blanket (Harvard Apparatus, Holliston, MA, U.S.A.). After the neck was incised in the anterior midline, the left external carotid artery was exposed, ligated with a 6–0 silk suture and dissected distally, and its branches were electrocoagulated; then the left internal carotid artery was isolated and separated from the vagus nerve. A 5–0 surgical monofilament nylon suture (Devis and Geck, Manati, Puerto Rico) blunted at the end was introduced into the left internal carotid artery through the external carotid artery stump and advanced 9 or 10 mm past the common carotid bifurcation. The skin was sutured within 10 minutes of incision. After recovery from anesthesia, mice were kept in a 20°C air-conditioned room. Regional cerebral blood flow (rCBF) was measured by laser-Doppler flowmetry (Moor Instruments, Devon, U.K.). The probe was positioned over the right hemisphere, 0.5 mm anterior and 2 mm lateral to the bregma. rCBF was monitored continuously for 5 minutes (Chuquet et al., 2002).
Infarct volume
Mice (n = 7 per condition) were anesthetized with C2H3Cl3O2 and decapitated 24 hours after the onset of ischemia. Brains were removed and 1-mm coronal sections immersed in 2% TTC (2,3,5-triphenyltetrazolium hydrochloride) in saline for 20 minutes at 37°C, then fixed for 2 hours in 4% paraformaldehyde (Bederson et al., 1986a). Infarct volume was determined using the NIH Image program.
Neurologic testing
Mice were evaluated by a blinded observer 24 hours after ischemia using a five-point scale, where 0 = normal motor function, 1 = contralateral torso and forelimb flexion on lifting the animal by the tail, 2 = circling to the contralateral side with normal posture at rest, 3 = leaning to the contralateral side at rest, and 4 = no spontaneous motor activity (Bederson et al., 1986b; Sun et al., 2003a).
Cell culture
Neuron-enriched (>95% microtubule-associated protein 2–immunopositive) cultures were prepared from the cerebral hemispheres of 16-day-old Charles River CD1 mouse embryos (Jin et al., 2000b), seeded at 3 × 105 cells per well on 24-well dishes precoated with poly-D-lysine, and grown in Eagle's minimal essential medium (Gibco BRL, Rockville, MD, U.S.A.) with 5% horse and 5% fetal bovine serum. Cultures were treated with 10-μmol/L cytosine arabinoside on day 6 and used on day 11. VegfB (rhVEGFB167, catalog #751-VE; R&D Systems; 100 ng/mL) was added to some cultures immediately before the onset of hypoxia. Hypoxia was induced by placing cultures in modular incubator chambers (Billups-Rothenberg, Del Mar, CA, U.S.A.) containing humidified 95% N2 and 5% CO2 for 16 hours at 37°C, and then returning them to normoxic conditions (humidified 95% air and 5% CO2) for another 8 hours. Control cultures were maintained under normoxic conditions for the entire 24-hour period. Hypoxic neuronal injury was measured by incubating cultures with 5 mg/mL of MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] at 37°C for 2 hours and measuring A570 nm in solubilized cells using a Cytofluor Series 4000 plate-reader (PerSeptive Biosystems, Framingham, MA, U.S.A.). Results were confirmed by trypan blue exclusion.
RESULTS
VegfB-knockout mice were bred in-house in 1:2:1 mendelian ratios of VegfB+/+:VegfB+/−:VegfB−/−. As reported previously, VegfB+/− and VegfB−/− mice were indistinguishable by observation from wild-type mice (Aase et al., 2001; Bellomo et al., 2000). A 247-bp fragment from the wild-type allele and a 74-bp fragment from the mutant allele were amplified. Reverse transcription PCR and Western blotting (Fig. 1) showed that VegfB mRNA and protein were detectable in VegfB+/+ and VegfB+/− mice, but not in VegfB−/− mice. Double-label immunohistochemistry in brains of VegfB+/+ mice showed that VegfB protein colocalized with the mature neuronal marker NeuN, the endothelial cell marker CD146, and the vascular smooth muscle cell marker α-actin (but not the astroglial marker glial fibrillary acidic protein), indicating that VegfB was expressed in neurons, endothelial cells, and smooth muscle cells (Fig. 2). The distribution of major cerebral surface vessels and the density of intracerebral vessels were similar in VegfB−/− and VegfB+/+ mice, as was the permeability of cerebral vessels to IgG (Fig. 3).
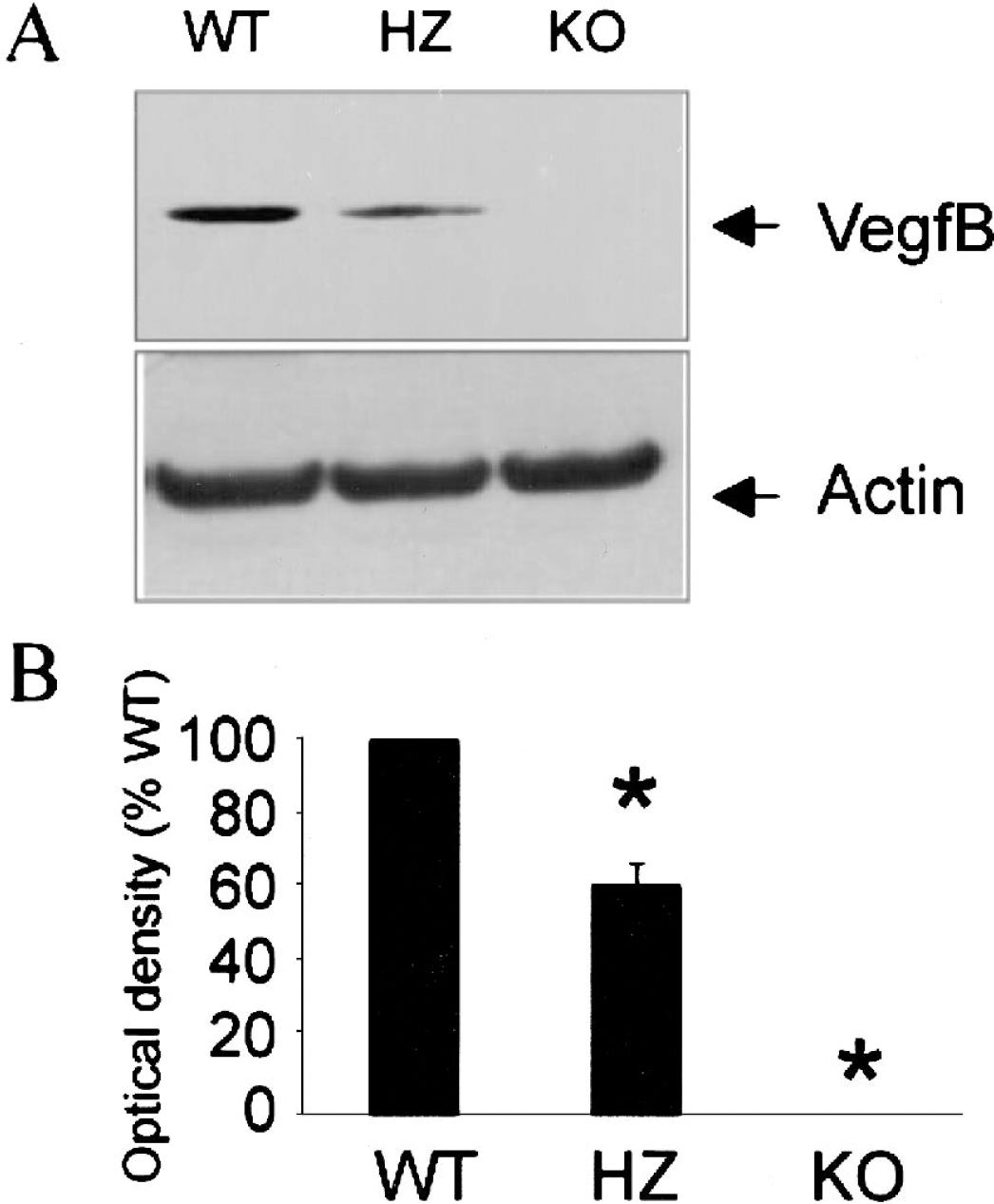
Western blot analysis of VegfB protein (Mr ≈ 19 kd) expression in mouse forebrain. (
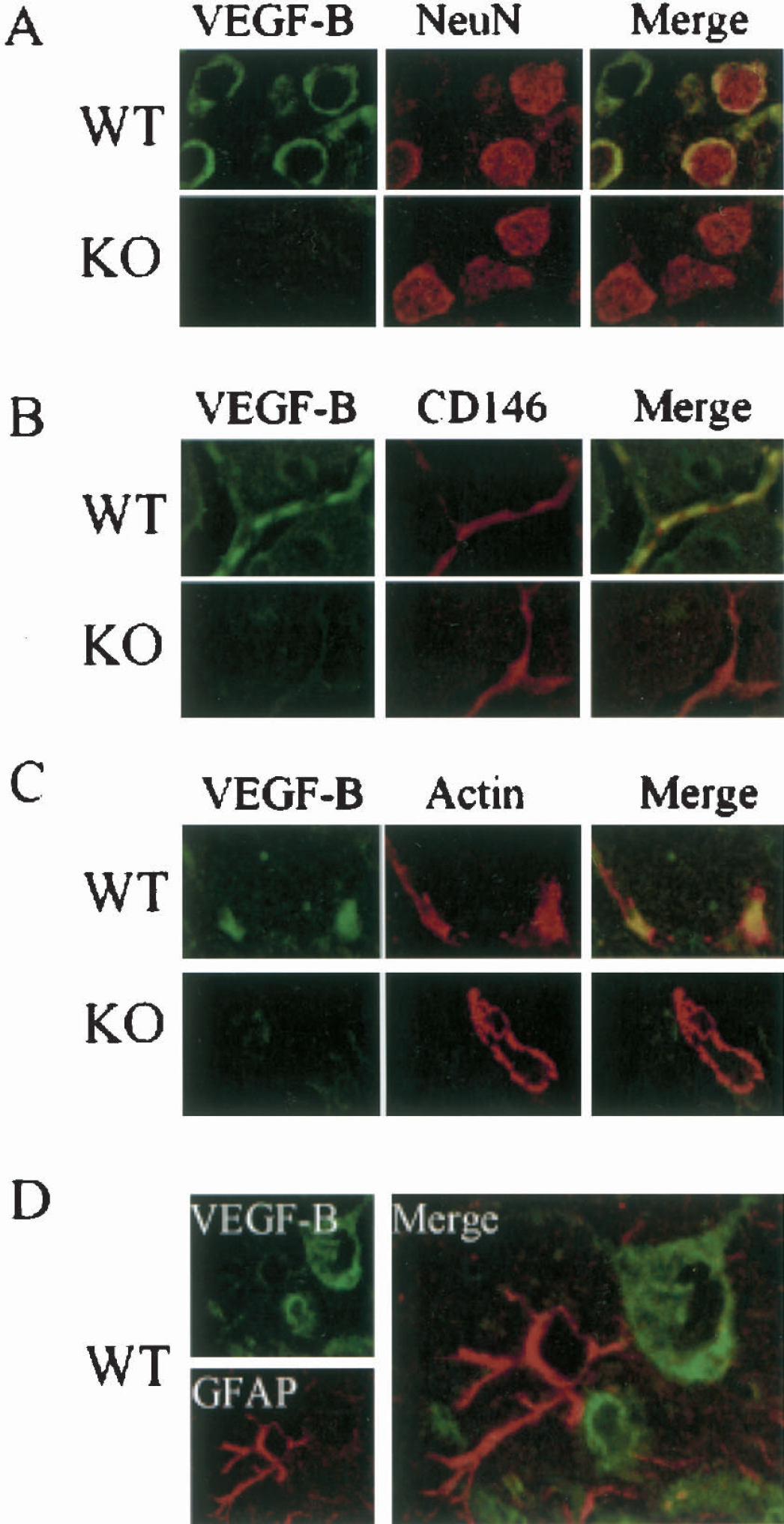
Immunocytochemistry of VegfB protein expression in cerebral cortex of wild-type (WT) and VegfB-knockout (KO) mice. Double-labeling shows colocalization in WT but not KO mice of VegfB with (
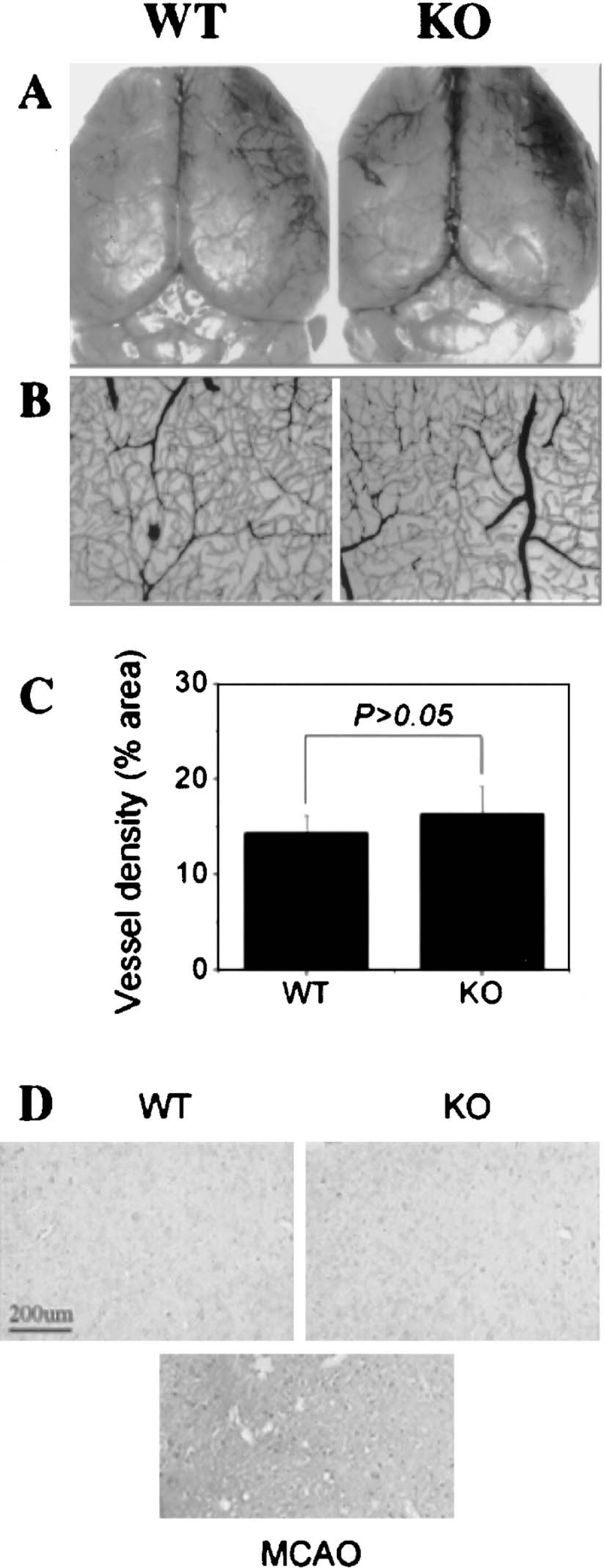
Cerebral vasculature in wild type (WT) and VegfB-knockout (KO) mice. The distribution of major cerebral surface vessels (
To investigate whether VegfB protects the brain from ischemia, MCAO was produced and infarct areas measured 24 hours later. As shown in Figs. 4A and 4B, infarct volume (determined by TTC staining, which delineates intact tissue) was increased approximately 40% in VegfB−/− compared with VegfB+/+ mice (P < 0.05); this was confirmed by hematoxylin staining (not shown). Neurologic scores, which reflect the severity of brain dysfunction after cerebral ischemia, were also more severely impaired at 24 hours in VegfB−/− compared with VegfB+/+ mice (Figure 4C). Baseline rCBF was unchanged in VegfB−/− mice (100% ± 3% of wild-type mice, n = 12). There were no statistically significant differences across groups in arterial blood pH (range, 7.31 to 7.37), partial pressure of O2 (range, 151 to 188 mm Hg) or CO2 (range, 36 to 40 mm Hg), or body temperature (range, 36.8 to 37.3°C), measured before or 30 minutes after the onset of MCAO.
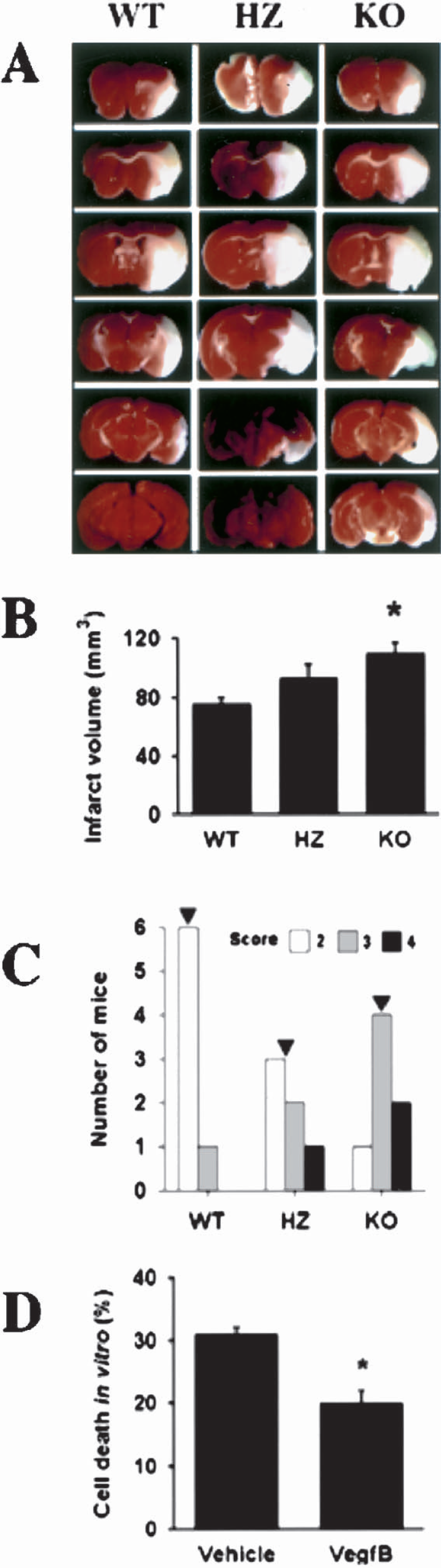
Effect of VegfB gene knockout on infarct volume and neurologic function after MCAO in vivo, and direct neuroprotective action of VegfB in vitro. (
VegfA is directly neuroprotective, and has been shown to protect cultured neurons from hypoxic injury (Jin et al., 2000b). To determine if VegfB is also directly neuroprotective, cultured murine cerebrocortical neurons were made hypoxic for 16 hours in the presence or absence of 100 ng/mL of VegfB, a concentration found in preliminary experiments to be maximally effective. Hypoxic injury was expressed as the percent decline in MTT absorbance in hypoxic compared with normoxic cultures. Hypoxia injured approximately 30% of cells in the absence of VegfB, but only approximately 20% of cells in its presence, consistent with a direct neuroprotective effect of VegfB (Fig. 4D). A VegfB-induced decrease in neuronal injury of similar magnitude (to 69% ± 5% of control, n = 3) was seen when cell viability was measured by trypan blue exclusion.
DISCUSSION
We found that the size of cerebral infarcts and the severity of neurologic deficits increased in mice lacking the VegfB gene. This implies that in wild-type mouse brain, VegfB is neuroprotective because it limits the extent of cerebral ischemic injury. In this respect, VegfB resembles VegfA. Although VegfA gene knockout results in embryonic lethality, topical, intravenous, or intraventricular administration of VegfA improves outcome from experimental stroke in rodents (Hayashi et al., 1998; Sun et al., 2003b; Zhang et al., 2000), whereas intraventricular administration of an anti-VEGF antibody increases infarct size (Bao et al., 1999).
Protection from cerebral ischemia by VegfA likely results from a direct effect on neurons that is mediated through Vegfr2/Flk1 receptors. VegfA exerts multiple effects in the ischemic brain, including stimulation of neurogenesis and angiogenesis, but a VegfA-induced decrease in infarct size is evident before these effects occur (Sun et al., 2003b). VegfA also protects cortical neurons from simulated ischemia in vitro, and this also involves an interaction with Vegfr2/Flk1 receptors, because it occurs in cells that lack Vegfr1/Flt1 receptors and it is not reproduced by the Vegfr1/Flt1 and Nrp1 ligand, Plgf (Jin et al., 2000c).
The mechanism through which VegfB reduces ischemic neuronal injury is less clear, but previous studies and our results provide clues. VegfB binds preferentially to Vegfr1/Flt1 (Olofsson et al., 1998) and Nrp1 (Makinen et al., 1999), and not to the Vegfr2/Flk1 receptor that appears to mediate the neuroprotective effects of VegfA. Both Vegfr1/Flt1 (Lennmyr et al., 1998) and Nrp1 (Kolodkin et al., 1997) are expressed on neurons, so VegfB could act directly at these sites. Our finding that VegfB protects cultured neurons from hypoxia is consistent with such an action, although VegfB might have other effects on neurons or act on additional cell types in vivo. For example, VegfA is not only neuroprotective, but also promotes angiogenesis and neurogenesis in the ischemic brain (Zhang et al., 2000; Sun et al., 2003b), and either or both of these processes could influence outcome after stroke. VegfB-knockout mice show more severe deficits after myocardial ischemia, including increased peak contracture, increased diastolic blood pressure, and slowed recovery of contractile function (Bellomo et al., 2000). This might suggest an additional mechanism for VegfB's protective action, such as an effect mediated through the vasculature.
If lack of VegfB produced anomalies of cerebral vasculature, this could provide a trivial explanation for its “neuroprotective” effect. VegfB-knockout mice have small hearts (Bellomo et al., 2000) and atrial conduction defects (Aase et al., 2001), but both cardiac (Bellomo et al., 2000) and retinal (Reichelt et al., 2003) blood vessels develop normally in these animals, and we found no abnormalities of cerebral vessels or rCBF in this study. Therefore, it seems unlikely that VegfB- knockout mice fare worse after stroke simply because of defects in, for example, collateral circulation.
The observation that VegfB-knockout mice exhibit more severe cerebral ischemic injury seems at odds with the considerable redundancy that appears to be built into VegfB signaling. Phenotypic abnormalities in unstressed VegfB-knockout mice are minimal (Bellomo et al., 2000), and Plgf interacts with the same receptors as VegfB, and so might be expected to substitute fully for it. Plgf's limited hypoxia-responsiveness cannot explain its failure to replace VegfB, because VegfB is also less hypoxia-responsive than, for example, VegfA (Yonekura et al., 1999). Moreover, both VegfB (Nag et al., 2002) and PlGF (Beck et al., 2002) levels increase after ischemia or related brain injury.
The present study implicates VegfB in the brain's response to ischemia, and establishes it as another likely endogenous neuroprotective factor. Several questions remain unanswered about the role of VegfB in cerebral ischemia, however. Is the protective effect of VegfB due solely to a direct neuroprotective action? Is VegfB preferentially induced in brain regions that survive ischemic insults? Is Vegfr1, Nrp1, or both required for protection by VegfB? Are the protective effects of VegfA and VegfB additive? Does Plgf also protect against cerebral ischemia? These issues will be important to address in future studies.