
Abstract
The aim of the present study was to develop an experimental paradigm for the study of serotonergic neurotransmission in humans using positron emission tomography and the 5-HT2A selective radioligand [18F]altanserin. [18F]altanserin studies were conducted in seven subjects using the bolus/infusion approach designed for attaining steady state in blood and brain 2 hours after the initial [18F]altanserin administration. Three hours after commencement of radiotracer administration, 0.25 mg/kg of the selective serotonin reuptake inhibitor, citalopram (Lundbeck, Valby, Denmark), was administered to all subjects as a constant infusion for 20 minutes. To reduce 5-HT1A–mediated autoinhibition of cortical 5-HT release, four of the seven subjects were pretreated with the partial 5-HT1A agonist pindolol for 3 days at an increasing oral dose (25 mg on the day of scanning). In each subject, the baseline condition (120 to 180 minutes) was compared with the stimulated condition (195 to 300 minutes). Despite a pronounced increase in plasma prolactin and two subjects reporting hot flushes compatible with an 5-HT–induced adverse effect, cortical [18F]altanserin binding was insensitive to the citalopram challenge, even after pindolol pretreatment. The biochemical and cellular events possibly affecting the unsuccessful translation of the citalopram/pindolol challenge into a change in 5-HT2A receptor binding of [18F]altanserin are discussed.
During the last decade several positron emission tomography (PET) studies and single photon emission computed tomography studies have demonstrated changes in radioligand binding upon pharmacologic challenge aiming at increasing the extracellular concentration of neurotransmitter (Laruelle, 2000). The technique has proven valuable in the study of the dopaminergic neurotransmission in schizophrenia (Breier et al., 1997; Laruelle et al., 1996) with possible implications for the understanding of the pathophysiology of the disease and the development of new treatment strategies (Carlsson et al., 1999). However, extending the concept to the study of other neuroreceptor systems has been difficult in terms of measuring a robust change in radioligand binding after pharmacologic challenge for the D1 receptor system (Abi-Dargham et al., 1999; Chou et al., 1999), the 5-HT1A receptor system (Abi-Dargham et al., 1997; Hume et al., 2001; Maeda et al., 2001), and the nicotinic acetylcholine receptor system (Fujita et al., 2003). In addition, increasing the extracellular concentration of dopamine only appears to affect the binding of benzamide ligands (such as IBZM and raclopride) to the dopamine D2 receptors whereas the binding of butyrophenone ligands (such as spiperone and NMSP) to the D2 receptors is unaffected (Laruelle, 2000). These findings emphasize the need for understanding the complex biochemical and cellular processes underlying neurotransmission studies with implications for future radiotracer development and experimental strategies.
The aim of the present study was to develop an experimental paradigm for the study of serotonergic neurotransmission in humans using PET and the 5-HT2A selective radioligand [18F]altanserin. The 5-HT2A receptor system is known to modulate mood and perception (Roth et al., 1998) and to mediate the effects of many commonly prescribed therapeutic drugs, including atypical antipsychotics and antidepressants (Kroeze et al., 2002; Meltzer et al., 1989). We used a combination of a selective serotonin reuptake inhibitor (SSRI), citalopram, and a partial 5-HT1A agonist, pindolol, to increase the extracellular concentration of 5-HT. The biochemical and cellular mechanisms possibly affecting the vulnerability of [18F]altanserin binding to an increase in extracellular 5-HT are discussed.
MATERIALS AND METHODS
Subjects
Studies were conducted in seven subjects (five women, two men) with a mean age of 29 years (range 23 to 35 years). All subjects claimed to be alcohol- and drug-free and had no history of neurologic or psychiatric disorders. Physical examinations, ECG, and rputine blood test results were normal in all subjects. All volunteers gave informed written consent. The ethical committee of Copenhagen and Frederiksberg approved the study (KF 02 to 058/99, 12 to 042/01, 12 to 131/01).
Radiochemistry
[18F]altanserin was synthesized as previously reported (Lemaire et al., 1991). The specific activity at the end of synthesis was 178 ± 51 GBq/mmol and the radiochemical purity was greater than 99%.
Positron emission tomography studies
Cannulas were inserted into both cubital veins for radiotracer administration and blood sampling, respectively. Subjects received a maximum of 3.7 MBq/kg body weight of [18F]altanserin. [18F]altanserin was administrated as a combination of a bolus injection and a continuous infusion to produce tracer steady state in tissue and blood. The bolus component was worth 1.75 hours of constant infusion (Pinborg et al., 2003). During constant infusion of [18F]altanserin, dynamic PET (23 frames of 8 minutes) was performed starting 2 hours after bolus injection. PET scanning was performed in a GE-Advance scanner (General Electric Medical Systems, Milwaukee, WI, U.S.A.) operating in three-dimensional acquisition mode with technical specification as previously described (DeGrado et al., 1994; Lewellen et al., 1996). Subjects were positioned with the orbitomeatal line parallel to the detectors ring planes and head movements were restricted with straps. To facilitate accurate repositioning, marks were placed along the orbitomeatal line and glabella and when needed, realignment was performed during the experiment with the help of laser lights. For correction of tissue attenuation, 10-minute transmission scans were performed before each PET session using retractable 68Ga/68Gepin sources. The transmission scans were corrected for tracer activity by a 5-minute emission scan performed in two-dimensional mode. Data were reconstructed into a sequence of 128 × 128 × 35 voxel matrices, each voxel measuring 2.0 × 2.0 × 4.25 mm, with software provided by the scanner manufacturer. A three-dimensional reprojection algorithm with a transaxial Hann filter (6 mm) and an axial ramp filter (8.5 mm) was applied. Corrections for deadtime, attenuation, and scatter were performed.
During PET scanning, 16 venous blood samples were drawn for determination of the total plasma radioactivity concentration and the plasma fraction of [18F]altanserin. The plasma fraction of native [18F]altanserin was determined in 9 of 16 venous plasma samples through high-performance liquid chromatography analysis as previously described (Pinborg et al., 2003). For time-points where high-performance liquid chromatography analysis was not performed, linear interpolation was done.
One hour after commencement of PET scanning, 0.25 mg/kg of citalopram (Lundbeck, Valby, Denmark) was administered to all subjects as a constant infusion for 20 minutes. A similar dose has been used in previous studies in normal volunteers (Kapitany et al., 1999; Seifritz et al., 1996) leading to an increase in plasma prolactin and plasma cortisol and with side effects (nausea, sweating, dizziness) staying within a tolerable range in all subjects. After citalopram administration, ECG was monitored online and blood pressure was monitored at approximately 15-minute intervals. The last four subjects were pretreated with pindolol (Gerard) at an increasing oral dose starting with 2.5 mg x 3 on day 1, 5 mg x 3 on day 2, 7.5 mg x 3 on day 3. On the day of the PET scanning (day 4) subjects received 7.5 mg at 8
Regions of interest definition
Magnetic resonance imaging was performed on a 1.5-T Vision scanner (Siemens, Erlangen, Germany). A total of 158 slices were acquired continuously in the sagittal plane using a three-dimensional MPRAGE sequence (echo time of 4 ms, repetition time of 11.4 ms, inhibition time of 100 ms, flip angle of 8 degrees, 256 × 256 image matrix, 1.14-mm slice thickness, and 1.21 × 1.21-mm in-plane resolution). PET images were aligned and the anatomical magnetic resonance imaging scans were coregistered to the individual averages of the aligned PET images as previously described (Pinborg et al., 2003). ROIs were delineated on the magnetic resonance imaging images and applied to the 23 aligned PET scans, thus generating time–activity curves for neocortical ROIs and cerebellum.
Derivation of the outcome parameter
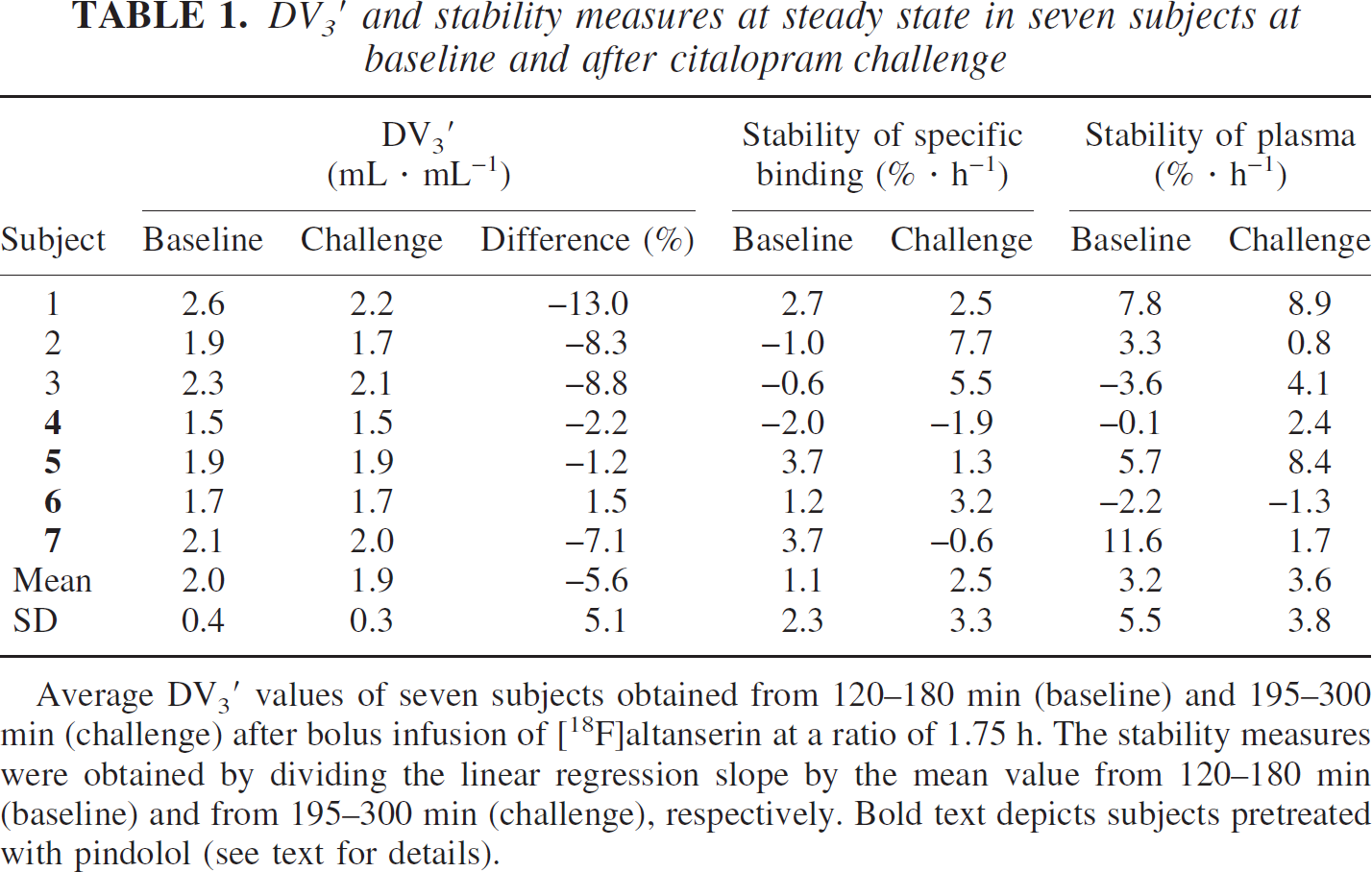
We used DV3′ as an outcome parameter (Pinborg et al., 2003):
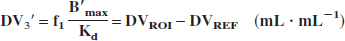
The distribution volume (DV) is defined as the ratio of the steady state tissue ligand concentration to the steady state plasma ligand concentration. DV3′ is calculated as the difference between the total distribution volume of [18F]altanserin in a region of interest (DVROI) and the distribution volume of [18F]altanserin in cerebellum (DVREF). The calculation of DV3′ presumes that DVREF has the same size as the distribution volume of free, nonspecifically bound and radiolabeled metabolites of [18F]altanserin in the region of interest. If the free fraction of [18F]altanserin in plasma (f1) is constant in time, DV3′ is a linear function of the ratio of the concentration of available 5-HT2A receptors, B′ max (nmol/L), to the equilibrium dissociation constant, Kd (nmol/L).
RESULTS
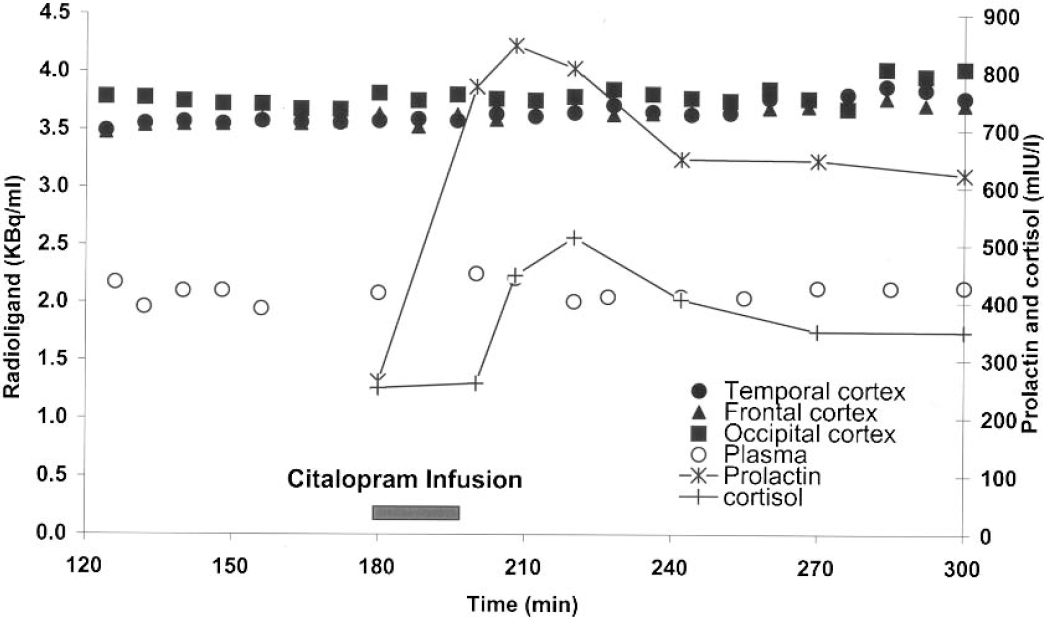
Time–activity curves of metabolite-corrected plasma and neocortical specific binding before and after citalopram challenge are shown in Fig. 1 (subject 1) and Fig. 2 (subject 6). The subject presented in Fig. 2 was pretreated with pindolol as described previously. It was not possible to demonstrate acute changes in plasma time–activity curves and brain time-activity curves upon acute citalopram challenge in any of the subjects studied including the subjects pretreated with pindolol. The plasma fraction of native [18F]altanserin was unaffected by citalopram administration and did not change with time. Baseline DV3′ was calculated from 120 to 180 minutes and challenge DV3′ was calculated from 195 to 300 minutes. Individual average neocortical DV3′ values are listed in Table 1. There was no significant difference between average neocortical baseline DV3,s and challenge DV3′s (P > 0.06, Wilcoxon matched pairs signed rank sum test). Likewise, no statistical differences were found in subdivisions of the neocortex. The stability of the specific binding of [18F]altanserin and plasma [18F]altanserin in the baseline and challenge situation was calculated by dividing the linear regression slope by the mean value from 120 to 180 minutes and 195 to 300 minutes, respectively. In subject 7, the measure of the stability of plasma from 120 to 180 minutes exceeded 10%/h (11.6%/h). A closer look at the plasma data revealed that the slope of the regression line were strongly influenced by a single outlier two standard deviations larger than the mean plasma [18F]altanserin concentration. However, overall there was no significant difference between the stability of specific binding at baseline compared to challenge (P = 0.66, Wilcoxon matched pairs signed rank sum test) and between the stability of plasma free parent compound at baseline compared to challenge (P > 0.55, Wilcoxon matched pairs signed rank sum test). Similarly, challenge DV3′ and measures of stability were calculated from 195 to 255 minutes and 255 to 300 minutes after bolus infusion of [18F]altanserin without showing significant differences compared to the baseline parameters.
Subject 1: time course of plasma parent compound and specific binding in cortical ROIs after bolus/infusion of [18F]altanserin. Specific binding was calculated as the difference between the cortical and cerebellar radioactivity concentrations. The cerebellar radioactivity concentration represents free and nonspecifically bound [18F]altanserin and radiolabeled metabolites. From 180 to 200 minutes, citalopram was administrated as a constant infusion (0.9 mg/min).
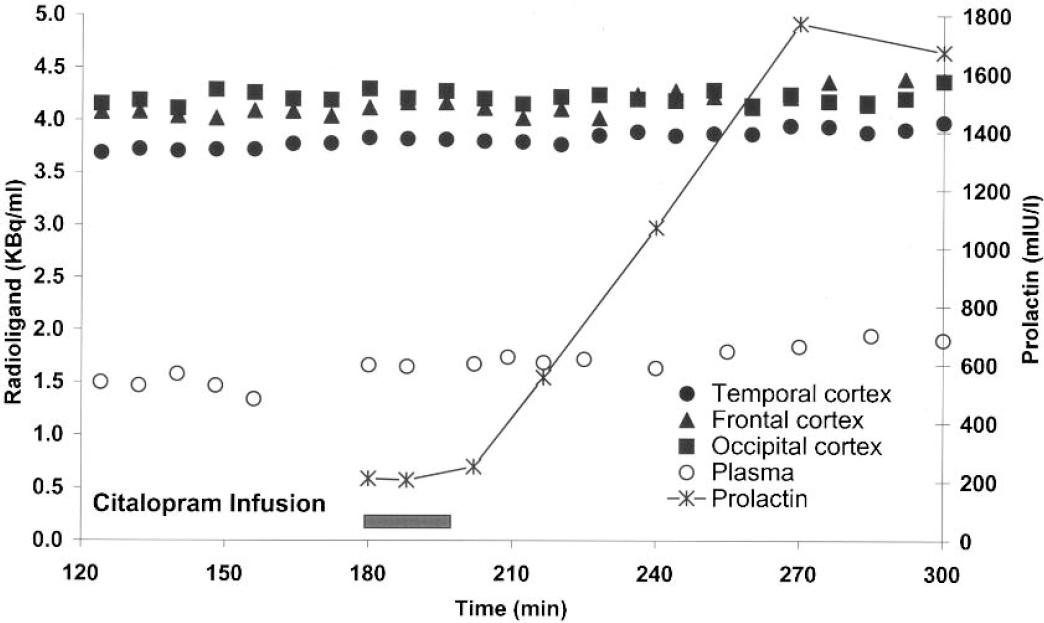
Subject 6: time course of plasma parent compound and specific binding in cortical ROIs after bolus/infusion of [18F]altanserin. Specific binding was calculated as the difference between the cortical and cerebellar radioactivity concentrations. The cerebellar radioactivity concentration represents free and nonspecifically bound [18F]altanserin and radiolabeled metabolites. From 180 to 199 minutes, citalopram was administrated as a constant infusion (0.7 mg/min). After 19 minutes of citalopram infusion (14 mg), this subject described a hot flush; citalopram infusion was stopped and the symptom disappeared within 2 minutes.
DV3′ and stability measures at steady state in seven subjects at baseline and after citalopram challenge
Average DV3′ values of seven subjects obtained from 120–180 min (baseline) and 195–300 min (challenge) after bolus infusion of [18F]altanserin at a ratio of 1.75 h. The stability measures were obtained by dividing the linear regression slope by the mean value from 120–180 min (baseline) and from 195–300 min (challenge), respectively. Bold text depicts subjects pretreated with pindolol (see text for details).
Table 2 shows the prolactin data of each subject. In contrast to the baseline plasma prolactin concentration (mean 208 mIU/L, SD = 35 mIU/L) there is a remarkable intersubject variation in the response to the weight corrected dose of intravenous citalopram—measured as the peak plasma prolactin concentration (mean = 683 mIU/L, SD = 532 mIU/L) or the area under the curve defined by the six plasma samples analyzed for prolactin from 180 to 300 minutes (mean = 29,560 mIU·L−1·min−1, SD = 28,032 mIU·L−1·min−1). There is no correlation between the percentage difference in DV3′ and peak plasma prolactin concentration (P > 0.29, Spearman correlation test) or the area under the curve (P > 0.48, Spearman correlation test). Subject 3 (not pretreated with pindolol) and subject 6 (pretreated with pindolol) described a hot flush after receiving 14 mg of citalopram. The heart frequency of subject 3 shortly dropped (ECG was consistent with sinus arrest), and a trained anesthesiologist considered these symptoms to be a vasovagal reaction to the unpleasant hot flush. Constant infusion of citalopram was stopped and symptoms were reversed within 2 minutes. Both subjects completed the experiment as planned—except for receiving only 75% (subject 3) and 97% (subject 6) of the scheduled citalopram dose. No other adverse reactions were seen. In subject 6, venous plasma samples were analyzed for plasma cortisol (Fig. 2). In the subjects pretreated with pindolol, the middle arterial blood pressure decreased by 14% (range, 4% to 23%) and the resting heart rate frequency decreased by 20% (range, 14% to 28%) as compared with values measured before pindolol treatment.
Prolactin response to the citalopram challenge
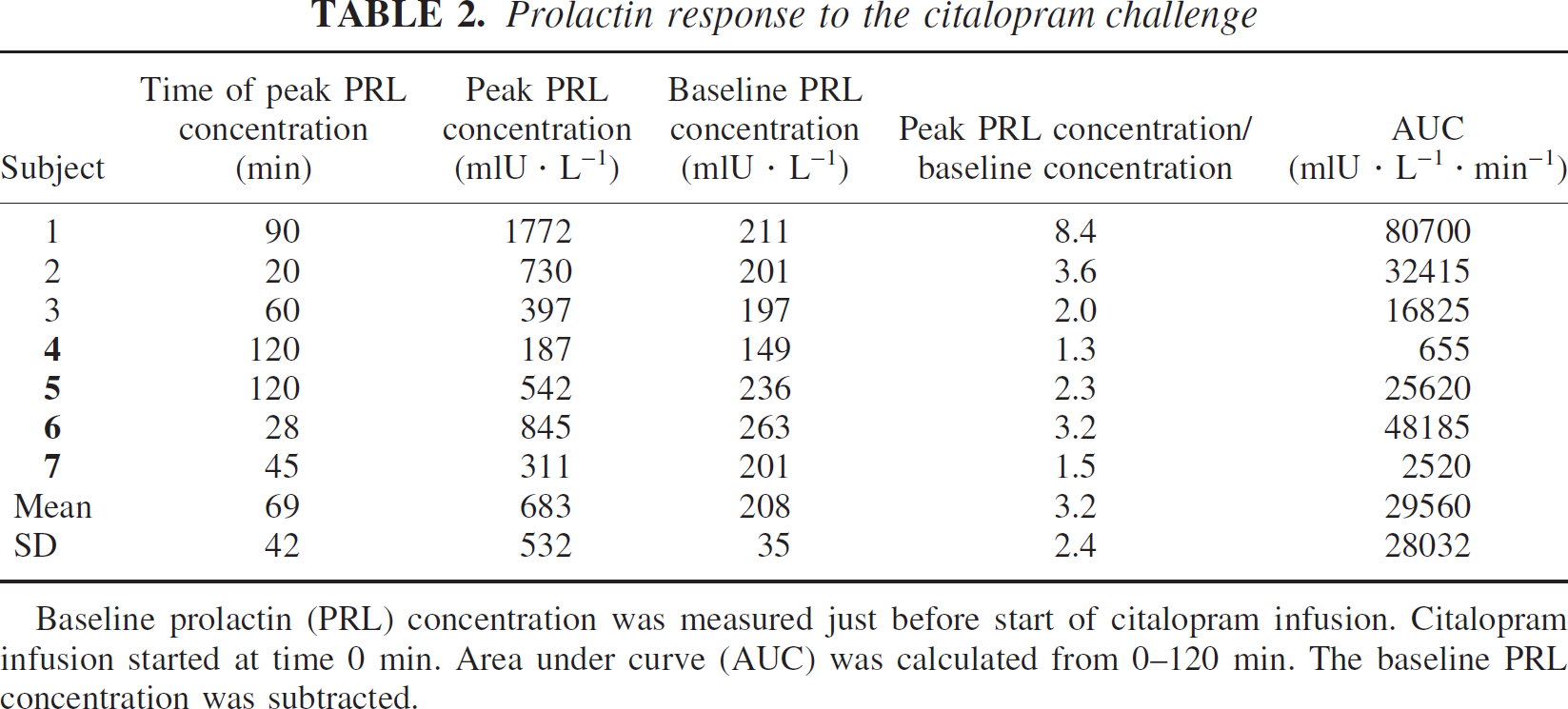
Baseline prolactin (PRL) concentration was measured just before start of citalopram infusion. Citalopram infusion started at time 0 min. Area under curve (AUC) was calculated from 0–120 min. The baseline PRL concentration was subtracted.
DISCUSSION
The aim of the present study was to develop an experimental paradigm for the study of serotonergic neurotransmission. We found, however, that [18F]altanserin binding to 5-HT2A receptors is insensitive to a citalopram challenge increasing extracellular 5-HT, and after pretreatment with pindolol. This negative finding rules out [18F]altanserin as a tracer sensitive for detection of changes in extracellular 5-HT, and it leads us to analyze the complex biochemical and cellular events translating the increase in extracellular neurotransmitter concentration into changes in radioligand binding to neuroreceptors.
Factors possibly influencing the success of a 5-HT2A receptor paradigm
During the experiment, the concentration of [18F]altanserin was kept constant at tracer doses. In an experiment where 5-HT competitively inhibit the binding of [18F]altanserin to the 5-HT2A receptor, the factors relating a change in the concentration of 5-HT at the 5-HT2A receptor to a change in the binding of [18F]altanserin to the 5-HT2A receptor is described as follows:
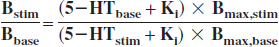
where Bstim and Bbase are the concentrations of [18F]altanserin bound to the 5-HT2A receptor after stimulation and at baseline, respectively; 5-HTbase and 5HTstim are the concentrations of 5-HT at the 5-HT2A receptor at baseline and after stimulation, respectively; Ki is the inhibition constant of 5-HT for [18F]altanserin; and Bmax, base and Bmax, stim are the concentrations of 5-HT2A receptors available for binding by 5-HT and [18F]altanserin at baseline and after stimulation, respectively. Kinetic analysis of [18F]altanserin bolus data (Biver et al., 1994) as well as ketanserin displacement studies (Pinborg et al., 2003) suggest that the [18F]altanserin rate constants for transfer from the bound compartment to the plasma compartment are sufficiently high to translate changes in the concentration of 5-HT or the number of available 5-HT2A receptors into changes in the time–activity curves of [18F]altanserin within the experimental time. In this study, the ratio of radioactivity in cortical ROIs to the radioactivity in cerebellum is approximately two throughout the experimental time. This corresponds to previous findings in a larger population (Pinborg et al., 2003). Thus, the specific signal is unlikely to be lost due to the contribution of radiolabeled metabolites. Theoretically, a decrease in specific radioligand binding could be obscured by a concomitant decrease in f1. We are not aware of any data, however, supporting that citalopram administration competitively decreases f1 for altanserin; in any instance, if a change in f1 had occurred, then it is likely that there would have been changes in the cerebellar DV with time, for which there was no evidence.
The 5-HT concentration
The concentration of neurotransmitter at the receptor is assumed to be largest in the synapse. For the dopamine D2 receptor system, the dopamine concentration varies from 6 to 200 nmol/L, with a temporal average synaptic dopamine concentration of approximately 100 nmol/L (May et al., 1988) compared with a dopamine concentration of 20 to 40 nmol/L in the extrasynaptic extracellular space (Church et al., 1987). In contrast to the D2 receptor system, 5-HT2A immunoreactivity appears to be absent in dendritic spines, which are the preferential site of synaptic contacts onto pyramidal cell dendrites. Instead, 5-HT2A receptors are primarily found in the dendritic shafts of cortical pyramidal cells (Jakab and Goldman-Rakic, 1998). In addition, the vast majority of 5-HT2A immunoreactive dendrites located some distance away from the 5-HT immunoreactive terminallike varicosities (Jansson et al., 1998), and 5-HT most likely induces its excitatory actions on neocortical pyramidal cells through extracellular diffusion (Jansson et al., 2001). Thus, compared with the D2 receptor system, augmentation of the 5-HT receptor system is likely to result in a smaller and probably more prolonged increase in 5-HT at the 5-HT2A receptor because of the spatial filtering of the response.
In the absence of 5-HT2A synaptic contacts, the extracellular concentration of 5-HT may be used as an approximation of the 5-HT concentration at the receptor. The baseline concentration of dialysate 5-HT in rat cerebral cortex has been measured to approximately 4 nmol/L (Hume et al., 2001). Administration of fenfluramine (10 mg/kg, intraperitoneally), an amphetamine analog, resulted in an approximately fivefold increase in extracellular 5-HT in medial prefrontal cortex (Hume et al., 2001). In the present study, citalopram was preferred to fenfluramine because of the reported association between the use of D-fenfluramine in combination with phentermine in the development of valvular heart defects (Connolly et al., 1997). Furthermore, fenfluramine and metabolites are substrates for the noradrenaline transporters and multiple 5-HT2 receptors subtypes (Rothman et al., 2003). Citalopram is a potent and selective serotonin reuptake inhibitor that, in contrast to tricyclic antidepressants, binds to cholinergic receptors only with negligible affinity (Millan et al., 2001; Milne and Goa, 1991). In rodents, administration of SSRIs has been reported to increase dialysate 5-HT in cortex approximately twofold (Arborelius et al., 1996; Gobert et al., 1997; Hervas et al., 2001; Invernizzi et al., 1992). The increase in cortical dialysate 5-HT with the use of SSRIs may be limited by 5-HT binding to somatodendritic 5-HT1A autoreceptors in the dorsal raphe nuclei leading to less pronounced 5-HT release in the frontal cortex (Artigas et al., 1996). The combined administration of SSRIs and 5HT1A receptor antagonists/partial agonists has been reported to further increase dialysate 5-HT in cortex to approximately threefold the baseline level (Arborelius et al., 1996; Gobert et al., 1997; Hervas et al., 2001; Invernizzi et al., 1992; Malagie et al., 1996; Sharp et al., 1997). Because selective 5HT1A blockers are not available for use in humans, we used the partial 5-HT1A agonist pindolol (Artigas et al., 1994) to reduce 5-HT1A mediated autoinhibition of cortical 5-HT release in four of the subjects. Based on the findings of previous PET studies of pindolol binding to human 5-HT1A receptors (Andree et al., 1999; Martinez et al., 2000; Rabiner et al., 2000), we believe that the pindolol treatment regimen used in this study is likely to result in a significant occupancy of dorsal raphe nuclei 5-HT1A receptors—probably around 40% to 50%. In this study, we used plasma prolactin and plasma cortisol as a marker of extracellular 5-HT concentration. The effect of SSRIs on plasma prolactin is mediated through 5-HT2A receptors (Curtis et al., 1995) and 5-HT1A receptors (Meltzer and Maes, 1994) in the dorsal raphe nucleus (Van de Kar and Bethea, 1982) and the hypothalamic paraventricular nucleus (Minamitani et al., 1987). As shown in Table 2, administration of intravenous citalopram resulted in an average increase in the peak plasma prolactin concentration to baseline plasma prolactin concentration ratio of 3.2 (range, 1.3 to 8.4). Notably, a large interindividual variation in both timing and peak prolactin concentration was found (Table 2). The exact relationship between the increase in plasma prolactin concentration and the increase in extracellular 5-HT concentration is unknown. However, the prolactin responses were brisk and pronounced in subjects 2 and 6, both of whom reported a hot flush compatible with an 5-HT–induced adverse effect (Berendsen 2000); part of the neuroendocrine response in these two patients could be a stress-response. Pretreatment with pindolol did not alter the stable time course of the [18F]altanserin time–activity curves (Fig. 2). Actually, the average prolactin response to citalopram was smaller in the group pretreated with pindolol, a finding consistent with the role of the 5-HT1A receptor in mediating the prolactin response (Meltzer and Maes, 1994).
The Ki value of 5-HT for [18F]altanserin
Whether a two- to threefold increase in extracellular 5-HT upon pharmacologic challenge will result in a measurable decrease in the binding of [18F]altanserin compared with baseline depends on the concentration of 5-HT compared with the Ki value of 5-HT for the [18F]altanserin binding sites according to Eq. 2. In our laboratory, a Ki value of 5-HT for [18F]altanserin of approximately 1,000 nmol/L has been measured using rat cortical membrane preparations. This value is comparable to the Ki values of 5-HT for [3H]ketanserin reported in the PDSP Ki database (http://kidb.cwru.edu/pdsp.php), which range from 70 to 3,000. Thus, in theory, an increase in the extracellular concentration of 5-HT from 4 to 12 nmol/L is unlikely to significantly affect the binding of [18F]altanserin to the 5-HT2A receptor. Two properties of [18F]altanserin potentially hamper its use for studies of 5-HT changes. Firstly, [18F]altanserin is a highly lipophilic molecule that can easily pass the blood–brain barrier. The plasma membrane and endosomal membrane are not expected to constitute quantitatively important diffusion barriers to [18F]altanserin, and the tracer will bind to the large (80% to 90%) intracellular pool of 5-HT2A receptors (Cornea-Hebert et al., 1999). By contrast, increased extracellular levels 5-HT are unlikely to be reflected accurately in the cytoplasmic compartment of the postsynaptic neurons because of a much lower lipophilicity. Secondly, [18F]altanserin is an antagonist at the 5-HT2A receptor, meaning that [18F]altanserin binds equally well to all affinity configurations of the 5-HT2A receptor; this is in contrast to 5-HT, which will bind only to the receptors configured in high-affinity state for agonist. Consequently, [18F]altanserin will bind to all 5-HT2A receptors, irrespective of their localization and configuration, whereas 5-HT probably binds primarily to 5-HT2A receptors located in the plasma membrane (10% to 20%), which is configured in high-affinity state for agonist binding (at best the majority).
The concentration of 5-HTM2A receptors available for binding
Although competition between neurotransmitter and radioligand at the receptor takes place, increasing evidence support the idea that agonist-induced desensitization of membrane-bound receptors is the major factor responsible for translating an increase in extracellular neurotransmitter concentration into a decline in radioligand binding (Laruelle 2000). This evidence stems from microdialysis studies where a temporal decoupling between changes in concentration of dialysate dopamine and changes in benzamide radioligand concentration has been shown with PET (Carson et al., 2001) and with single photon emission computed tomography (Laruelle et al., 1997). Further, in vitro studies have shown a decrease in [3H]raclopride Bmax upon amphetamine stimulation (Ross and Jackson, 1989; Sun et al., 2003). For the 5-HT2A receptor system, rapid receptor internalization (within 5 minutes) after either agonist or antagonist exposure has been shown in NIH 3T3 cells (Berry et al., 1996). However, because 80% to 90% of the 5-HT2A receptors are present in the intracellular compartment, agonist-induced receptor internalization is not likely to influence overall [18F]altanserin binding to any large extent. If [18F]altanserin binds equally well to the membrane-bound and intracellular pool of 5-HT2A receptors, a paradoxical increase in the binding of [18F]altanserin would be expected because fewer membrane-bound 5-HT2A receptors would be available for binding of 5-HT in competition with [18F]altanserin.
Thus, even though the citalopram/pindolol augmentation of the 5-HT receptor system via competitive and noncompetitive mechanisms may induce a change in the binding of [18F]altanserin to plasma membrane-bound 5-HT2A receptors, this signal may be to weak for detection.
Previous studies on 5HT2A neurotransmission
In a recent PET study, Larisch et al. (2003) reported a 14% decrease in cortical [18F]altanserin binding potential as compared with baseline values after the administration of 25 mg of intravenous clomipramine in 11 patients with a history of recurrent major depression (Larisch et al., 2003). The authors concluded that this decrease was caused by an increase in the 5-HT level leading to the lowered binding potential of [18F]altanserin. This interpretation is not in line with the biochemical and cellular factors possibly influencing the success of 5-HT neurotransmission studies using [18F]altanserin, as discussed previously. There may be several reasons for the discrepancy between their data and our results. Firstly, clomipramine is not a selective inhibitor of the serotonin transporter: it also binds to the norepinephrine transporter (Ki = 53.7 nmol/L), the 5-HT2A receptor (Ki = 35.5 nmol/L), the 5-HT2C receptor (Ki = 64.6 nmol/L), and the α-adrenoceptors (Ki = 3.2) (Millan et al., 2001). Although it binds to the 5-HT2C receptor with a low affinity (Ki = 617 nmol/L), citalopram binds to the other sites with Ki values > 10,000 nmol/L (Millan et al., 2001). In contrast to 5-HT, clomipramine is likely to compete with [18F]altanserin at all 5-HT2A binding sites and may, therefore, induce a decline in the [18F]altanserin binding. Finally, Larisch et al.‘s data were collected on two separate days—with and without clomipramine administration. Thus, data are susceptible to test–retest variability in the order of at least 10% as previously described (Smith et al., 1998). In five of the subjects in the Larisch et al.‘s study there was more than 20% difference in the cerebellar distribution volume in the baseline and stimulated situation. Because the cerebellar 5-HT2A receptor density is very small (Pazos et al., 1987; Schotte et al., 1983), and because PET studies with blocking doses of ketanserin failed to show any displacing effect on cerebellar [18F]altanserin binding (Pinborg et al., 2003), the difference in cerebellar distribution volume most likely reflects test–retest variability. In our study, the baseline and stimulated situation were defined during the same scanning session, thereby minimizing the test–retest variability (Pinborg et al., 2003).
Our data are supported by in vivo studies in anesthetized animals that failed to show changes in radioligand binding to 5-HT2A receptors upon pharmacologic challenges aimed at increasing extracellular 5-HT. Rice et al. (2001) reported that the binding of [3H]N-methylspiperone to cortical 5-HT2A in mice was unaffected by the 5-HT releaser, p-chloroamphetamine. Staley et al. (2001) reported that the binding of [18F]deuteroaltanserin to cortical 5-HT2A receptors in baboons was unaffected by 1.5 mg/kg IV fenfluramine.
Future neurotransmission studies of the 5-HT2A receptor system
The paradigm presented here with intravenous citalopram challenge worked well and is suitable for future neurotransmission studies of the 5-HT2A receptor system with other radioligands. A robust increase in neuroendocrine response was observed, and in line with this finding two subjects described mild and easily reversed side effects possibly related to the increase in extracellular 5-HT concentration upon inhibition of the serotonin transporter. The pindolol regime described in this study may be safely used to reduce 5-HT1A induced autoinhibition of 5-HT release.
Based on the biochemical and morphologic characteristics of the 5-HT2A receptor system—especially the fact that the majority of receptors are located intracellularly and only a minority of receptors are configured in high-affinity state for agonist binding—it seems unlikely that 5-HT2A receptor antagonist radioligands will prove successful in future neurotransmission studies of the 5-HT2A receptor system. An alternative strategy is to use full or partial agonist radioligands because they primarily bind to the same G-protein coupled receptor sites as 5-HT. The use of agonist radioligands could be hampered by a low target-to-background ratio because (partial) agonist ligands will bind only to a minor subpopulation of the 5-HT2A receptors compared with [18F]altanserin. Thus, = to increase the target to background ratio, the optimal (partial) agonist radioligand should posses a relatively high affinity (1/Kd) in combination with a low logP without hampering quantification and blood–brain barrier permeability.
Footnotes
Acknowledgments:
The authors thank Karin Stahr and the staff at the PET Center at Rigshospitalet, Copenhagen for expert technical assistance, the department of clinical biochemistry for conducting prolactin and cortisol analysis, and the John and Birthe Meyer Foundation for the donation of the cyclotron and PET scanner.