
Abstract
Positron emission tomography (PET) has become indispensable in the quantification of target engagement by brain targeting medications. The relationship between the drug plasma concentration (or drug dose administered) and target occupancy determined during a PET occupancy study has provided valuable information for the assessment of novel pharmaceuticals in the early phases of drug development. Such information is also critical for the understanding of the mechanisms of action and side-effect profile of approved medication commonly used in the clinic. Occupancy studies conducted following repeated drug dosing (RD) can produce systematic differences from those conducted following single drug dose (SD), differences that have not been adequately explored. We have hypothesised that when differences are observed between RD and SD studies, they are related to changes in target density induced by repeated drug accumulation. We have developed a modified occupancy model to account for potential changes in target density and tested it on a sample dataset. We found that target upregulation can parsimoniously explain the differences in drug affinity estimated in SD and RD studies. Our findings have implications for the interpretation of RD occupancy data in the literature and the relationship between specific target occupancy levels and drug efficacy and tolerability.
Introduction
Positron emission tomography (PET) has become an indispensable tool in the evaluation of medications for central nervous system (CNS) disorders, as it provides the only practical method to quantify drug occupancy of CNS targets in humans in vivo. These, so called “occupancy studies” have provided valuable insights into the pharmacology of medications used for the treatment of neurological and psychiatric illness 1 and have become an indispensable part of the process in the development of novel medications for CNS disorders.2,3
The design of occupancy studies involves the quantification of target availability of a particular receptor (or an enzyme, transporter, or another molecular target) using a PET ligand with properties that allow robust quantification of the relevant molecular target. Having obtained a baseline PET scan in a study participant, a dose of the drug under investigation is administered, and the availability of the molecular target is quantified again in a post-dose PET scan. The target occupancy is then calculated as the fractional reduction in the availability of molecular target to the binding of the PET radioligand. Improvements in this basic design have been developed to allow for the evaluation of the change in occupancy over time using adaptive study designs,4,5 however all the studies to date require the assumption that the total expression of the molecular target (or
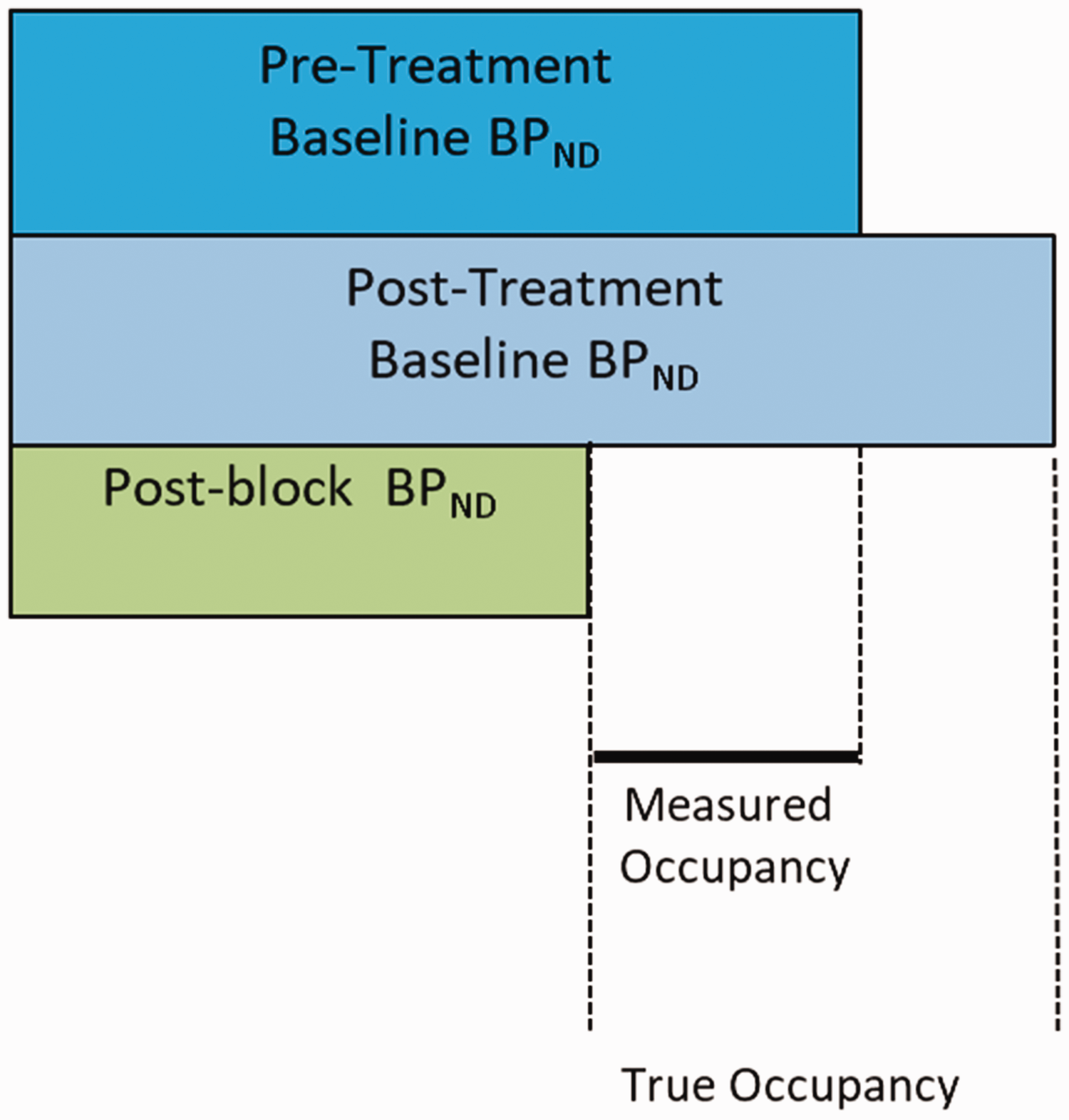
Effect of changes in baseline target availability on occupancy estimates. BPND - binding potential, a parameter often used in PET studies to represent target availability. As the true post-treatment target availability (in the absence of drug), represented here as “Post-treatment Baseline BPND” cannot be measured, the target occupancy is usually calculated as the fractional change in BPND from “Pre-Treatment Baseline BPND” to the “Post-Treatment Baseline BPND”. However, if the “Post-Treatment Baseline BPND” is not equal to the “Pre-Treatment Baseline BPND” due to factors such as target up- or down-regulation, then errors in the estimation of the true target occupancy will occur.
In this manuscript we have examined the biases induced by the violation of this assumption, and the consequences this has for the biological interpretation of occupancy study results. Violation of the assumption of unchanged target expression can occur in the context of an occupancy study that involves the administration of multiple doses of the investigational drug between the baseline and post-dose examination. A principle of pharmacology holds that an engagement of a target will lead to an upregulation of target expression in the case of an antagonist or a downregulation in the case of an agonist. The magnitude of up- or down-regulation will be specific for a particular neurotransmitter system and amongst other factors will depend on both the magnitude and the duration of target occupancy. Hence, an occupancy study that seeks to measure occupancy of a drug at steady state can lead to a significant bias. Any such bias will lead to erroneous estimation of the levels of occupancy required to produce clinical effects by established therapies. In addition, any such estimates of required receptor occupancy that are used during the development of novel pharmaceuticals will also suffer from such bias. Understanding the potential effects of such biases will help clarify conflicting data from past studies as well as help optimize the development of future medications.
Methods
General considerations
Under tracer conditions the binding of a radioligand (RL) to a molecular target can be quantified in terms of its binding potential with reference to the non-displaceable compartment (BPND
6
)
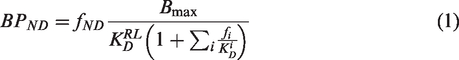
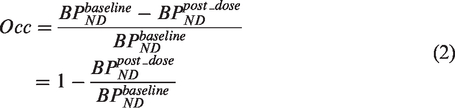
The fractional occupancy (Occ) of a target by competing species can then be calculated as
Assuming target occupancy by endogenous ligands is negligible and that there is a single competing drug, substitution of equation (1) into equation (2) provides a relationship between the affinity constant of an exogenously administered drug (
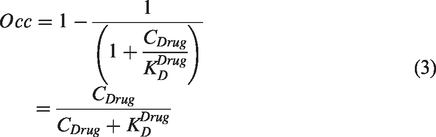
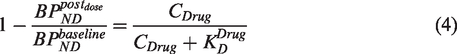
However, if drug administration results in an upregulation of the molecular target, equation (4) would lead to a biased result, and should therefore be modified accordingly. Such a situation can arise when the drug is administered for a prolonged period, and target upregulation (in the case of an antagonist drug) or downregulation (in the case of an agonist drug) has occurred. In such situations, the term
Thus, if a change in the target concentration (Bmax) is seen, with no change in the target affinity for either the drug (
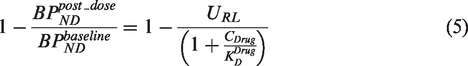
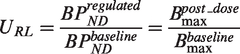
Equation (5) can be re-arranged to yield:
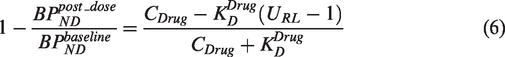
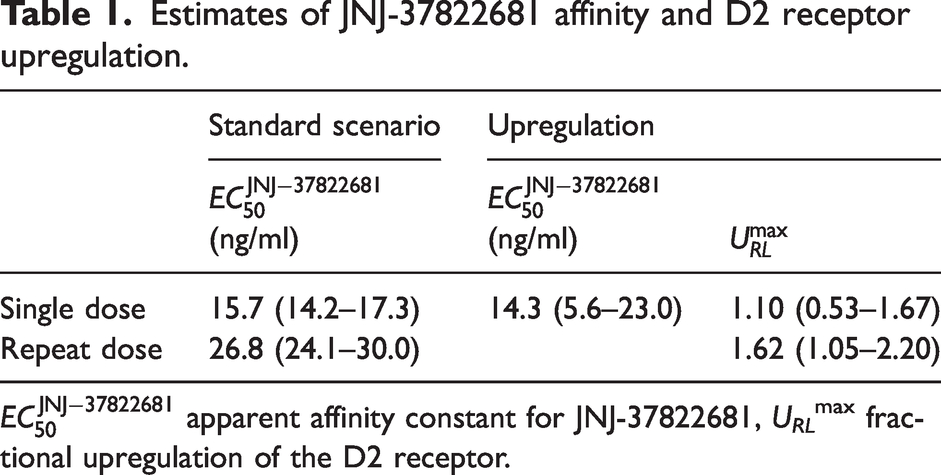
A comparison of equation (6) with equation (4) indicates that the application of a standard occupancy model in a situation where the target density has been upregulated following drug administration, will result in a measured occupancy that will underestimate the true occupancy in the case of target upregulation (URL > 1) or overestimate the true occupancy in the case of target downregulation (URL < 1).
Equation (6) describes the adjustment required for an equation correctly estimating target occupancy following a change in target Bmax, however, we cannot assume that URL is a constant for different drug doses. We do not have a practical method to measure the relationship between drug dose and the magnitude of URL. We have therefore made the explicit assumption that the magnitude of URL will be proportional to the degree of occupancy achieved by the blocking drug and modifying (6) accordingly provides:
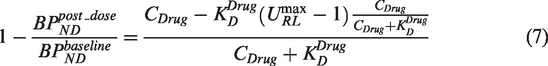
Evaluation of dopamine D2 receptor occupancy following repeat-dose administration
Experimental data for the D2 receptor system were obtained from publications evaluating the occupancy of a novel antipsychotic, JNJ-37822681 following both single dose (SD)
8
and repeat dose (RD)
9
administration. These data were chosen as they were derived from studies conducted by the same group, using consistent methodology, and provide plasma concentrations associated with individual subject occupancy for each of the subjects examined. The pre-scan plasma concentration of JNJ-37822681 and the measured D2R occupancy (derived using equation (2)) for both the SD and RD experiments were derived from Figure 2 of
9
Both SD and RD data were then fitted individually to equation (3) to determine the
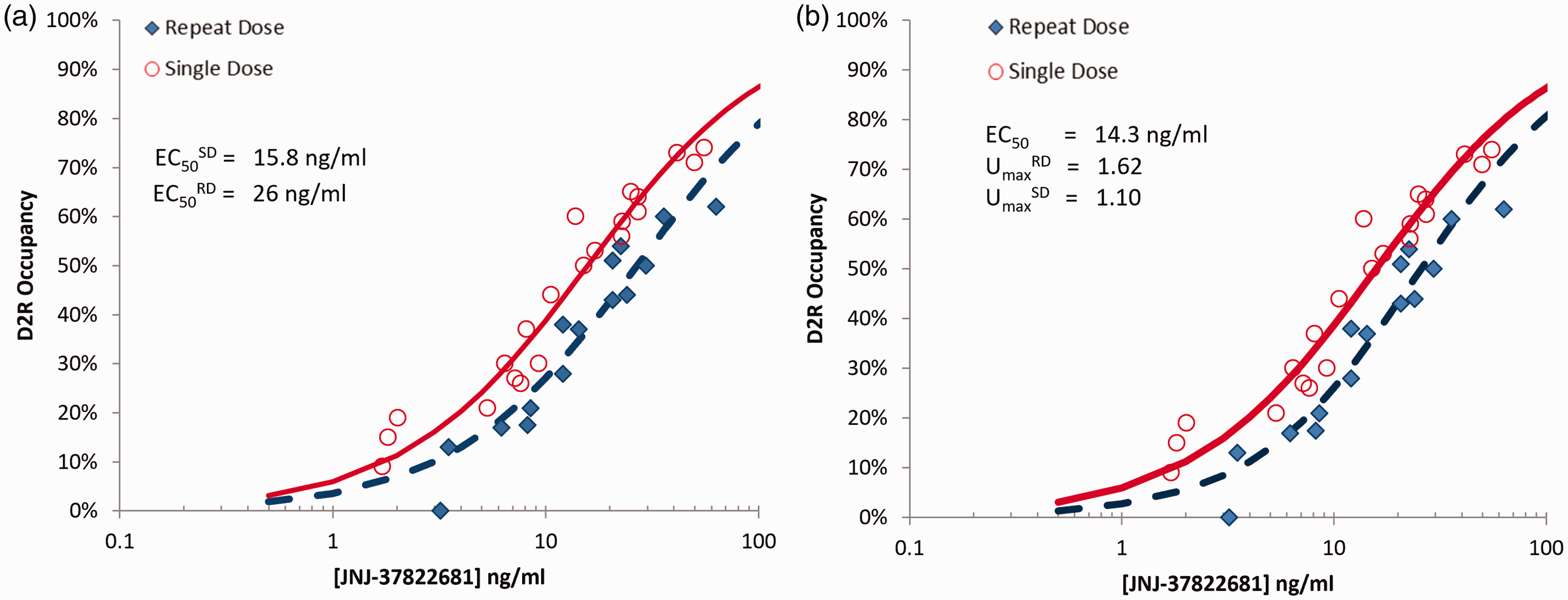
Data fits of single dose and repeat dose occupancy data to JNJ-37822681 plasma concentration. Occupancy data was estimated from [11C]raclopride PET data in the putamen, collected from healthy volunteers at baseline and following the administration of either a single oral dose (2–20 mg) of JNJ-37822681 (N = 16), or at steady state following twice daily dosing of 10 mg of JNJ-37822681 for 6 days (N = 15). 9 a) independent fits b) combined data fit accounting for potential upregulation of the D2 receptors. EC50 - plasma concentration leading to 50% target occupancy, UmaxRD - maximal fractional upregulation following repeat dosing, UmaxSD - maximal fractional upregulation following single dosing.
Results
Separate fitting of these data to equation (3), provided
A simultaneous fit of all the data (both SD and RD) data to (7), with the assumption that the
Estimates of JNJ-37822681 affinity and D2 receptor upregulation.
Discussion
In this manuscript we have provided a biologically plausible explanation for the apparent discrepancy that can be observed in occupancy estimates between single dose and repeat dose occupancy studies. We hypothesised that repeat dose studies can result in changes in target expression that lead to errors in the estimation of target occupancy and have derived a model and a modified occupancy equation to describe it. While we focused on receptor upregulation consequent to extended antagonist administration, the concept and equations apply equally well to target downregulation consequent to extended exposure to agonists. We have explored an example of D2R occupancy by a novel antagonist and have obtained results that parsimoniously and consistently describe target occupancy following both single dose and repeat dose occupancy experiments.
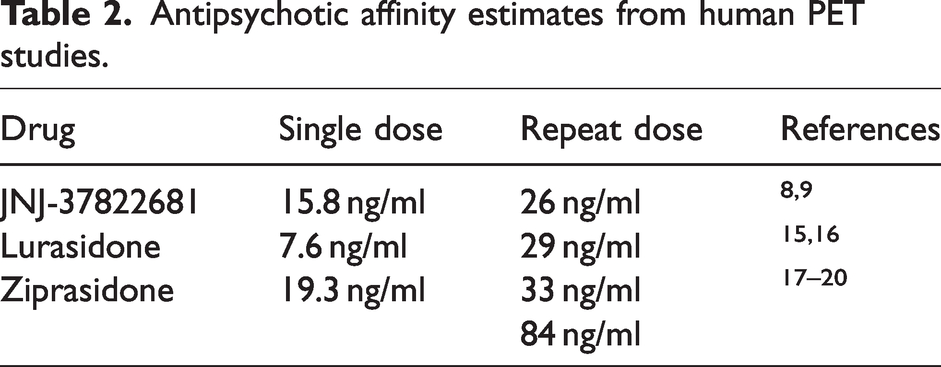
Human PET studies consistently report discrepancies in the relationship between plasma concentration of an antipsychotic and its occupancy of the brain D2R following single-dose (SD) and repeat-dose (RD) administration (see Table 2). A direct comparison of SD and RD data is complicated by the use of different PET ligands and quantification methods; however, a consistent finding is that antipsychotics display higher apparent affinity under SD administration paradigms than under RD administration. We have demonstrated that such differences can be parsimoniously explained by upregulation of target density following RD administration. Upregulation of target density following repeated administration of an antagonist, as well as downregulation of target density following repeated administration of an agonist are well recognized but have typically been neglected in the analysis of PET occupancy data. In particular, the estimation of D2R occupancy by antipsychotics in patients with schizophrenia has typically been performed by comparing the binding of D2R PET radioligands such as [11C]raclopride in healthy volunteers and untreated patients, to that of patients on chronic medication.10 –14 Such an analysis ignores any changes in receptor density and therefore will underestimate the true occupancy if a significant upregulation of D2R has occurred.
Antipsychotic affinity estimates from human PET studies.
The quantification of receptor upregulation in the living human brain is technically challenging, and hence has been typically examined in rodent ex vivo studies. Results of studies quantifying the upregulation of D2R in the rodent brain, following repeated antipsychotic administration over periods of 2–4 weeks, are summarized in Table 3. These data do not provide sufficient information to estimate the relationship between the magnitude of target occupancy and the degree of upregulation, nor to assess the time-course of upregulation. Nevertheless, rodent ex vivo experiments demonstrate that D2R upregulation of the order of 125–185% can be expected following RD administration of commonly used antipsychotics.
D2 receptor upregulation measured in the rodent brain.
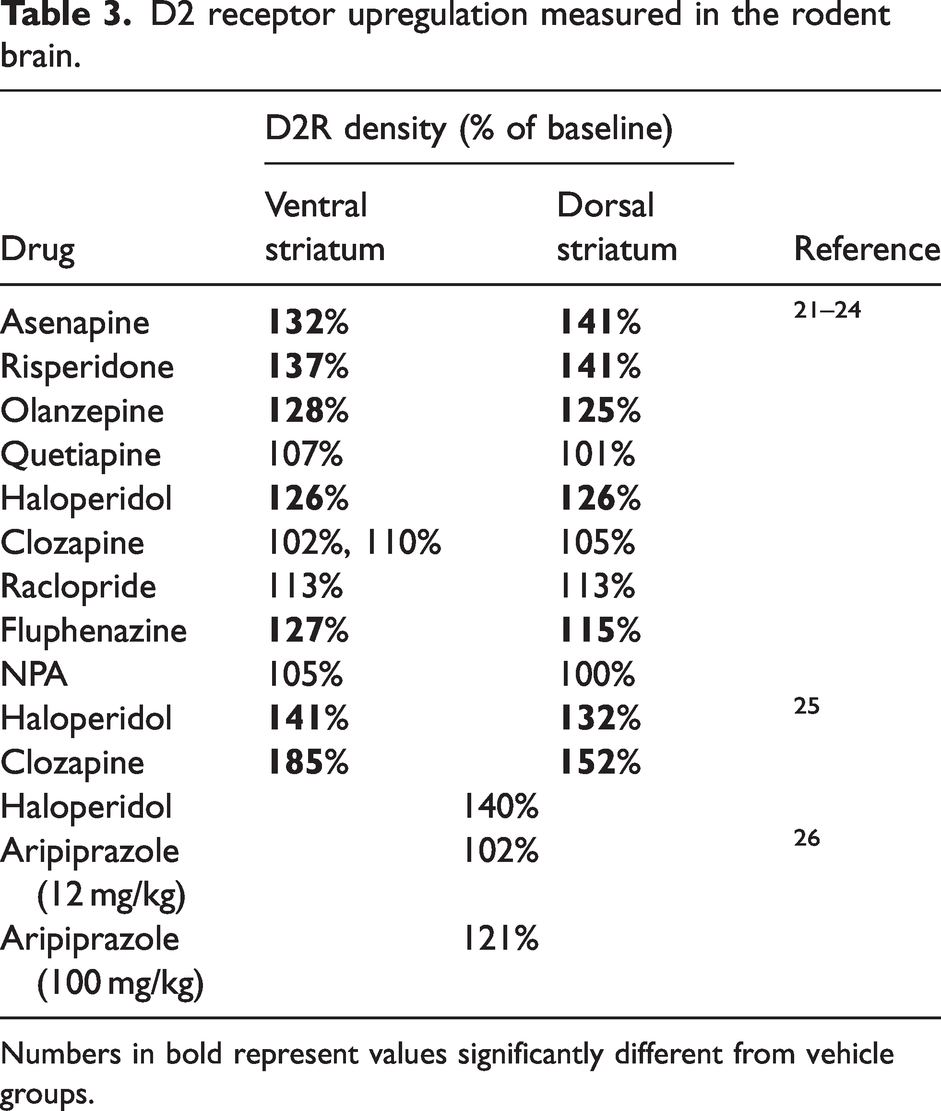
Numbers in bold represent values significantly different from vehicle groups.
If we assume that a similar level of upregulation would be observed in the human brain, then the commonly assumed target for clinically efficacious D2R occupancy by antipsychotics (of ∼65–80% as measured in RD PET studies) is in fact erroneously low, and the true range may be closer to 80–90%. While such an error may not be problematic for existing antipsychotics (for which the therapeutic doses have been estimated empirically based on clinical data), it may be of significant concern for drug developers who are aiming for a specific target occupancy to determine the therapeutic dose of a novel compound for clinical trials. If investigation of the magnitude and time-course of upregulation is of interest, our methodology provides an opportunity to investigate this process in the human brain. Once an in vivo affinity estimate (EC50), is obtained in a single dose occupancy study, various repeat-dosing scenarios can be examined to determine the apparent occupancy and equation (5) together with the single dose EC50 can be used to estimate the upregulation term (URL).
In the examples above we assumed that the affinity of the target for the radioligand and the blocking drug remains unaffected. While little evidence exists for a change in D2R affinity following chronic blockade, we cannot conclusively exclude changes in affinity from contributing to the differences seen in apparent drug affinity between single and repeat dose situations. However, the parsimonious explanation is one where a change in D2R number is the predominant factor.
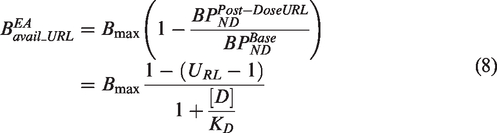
It may be argued that in the case of an antagonist drug, the important parameter is not the degree of occupancy of the receptor by the blocking drug, but rather its complementary quantity (1-Occupancy) which represents the number of receptors available for the action of the endogenous agonist (EA). In such a case, the error in the estimated occupancy when upregulation has occurred is not important, as the number of available receptors is still estimated correctly. However, the aim of an occupancy study is not just to describe the number of available receptors available for the EA at each post-dose PET scan, but to derive a general model that would enable the prediction of the number of receptors available for the action of the EA (BEAavail) at any drug plasma concentration [D] that may be used in future clinical efficacy trials. In the case where upregulation occurs, omission of the upregulation factor URL will result in an error in the estimation of BEAavail as can be seen from equation (8) below.
Changes in target affinity and endogenous neurotransmitter concentration
A change in target density between baseline and post-dose assessments is only one example of a situation where standard occupancy assessment will provide a biased estimate. A similar error will occur when the affinity of the target for the drug (D) and/or the radioligand (RL) changes following drug administration, such as in the case of targets that undergo allosteric modulation. An example is an enzyme such as phosphodiesterase (PDE) where the affinity of some PDE isoforms is modified by the binding of cyclic nucleotides (cN) to GAF domains on the enzyme.
27
As the PDE catabolise cN’s, the cellular concentration of the cN will be increased following a blockade of the PDE catalytic site, and hence the degree of allosteric modulation of the PDE, will be proportional to the level of blockade of the PDE catalytic site. The effects of such a change in affinity have been seen in vivo using PET.
28
Allosteric modulation will change the shape of the catalytic binding pocket, and the affinity of both the radioligand and the drug will be changed from baseline. If we make a simplifying assumption that such an alteration will change affinity for the drug and the radioligand to a similar degree measured occupancy will be described by (9).
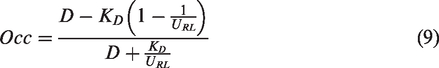
Another process that can lead to a change in the baseline binding potential (BPND) is a change in the concentration of the endogenous ligand (EL, which can be a neurotransmitter or another endogenous molecule interacting with the molecular target of the radioligand). Any drug treatment that leads to changes in the synthesis or release of EL, can lead to a change in the baseline BPND. Such effects can be difficult to detect and correct due to the lack of practical methods to quantify changes in EL in the brain in vivo. Most PET radioligands are relatively insensitive to changes in EL, and the ones that are known to be sensitive (such as [11C]raclopride, [11C]PHNO or [11C]Cimbi-36) demonstrate changes in baseline BPND of 15–25% at most, following the administration of powerful releasing agents such as D-amphetamine.29,30 While such changes can result in a bias in the estimation of an occupancy of an administered drug, the magnitude of this bias will be relatively small.
In conclusion, we have provided a biologically plausible model to explain the discrepancies in drug affinity seen between PET occupancy studies conducted following single dosing and repeat dosing regimens. The biases in the relationship between blood plasma concentration and levels of target occupancy, induced by the conduct of repeat dosing occupancy studies may lead to errors in assessing the relationship between clinical features of a drug and levels target occupancy. Single dose occupancy studies, when combined with time-occupancy assessment, 4 provide the best estimates of true target occupancy. Repeat dose occupancy data should be treated with caution and the effects of target up- or down-regulation should be considered.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: All work was funded by Invicro.
Acknowledgements
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: EAR and RNG are full-time employees of Invicro.
Authors’ contributions
EAR and RNG conceived and performed the work described, wrote this manuscript and approved the final version.