
Abstract
In this study, using both in vivo and in vitro ischemia models, the authors investigated the impact of brain ischemia on the biosynthesis of a key neuropeptide-processing enzyme, carboxypeptidase E (CPE). The response to brain ischemia of animals that lacked an active CPE was also examined. Combined in situ hybridization and immunocytochemical analyses for CPE showed reciprocal changes of CPE mRNA and protein, respectively, in the same cortical cells in rat brains after focal cerebral ischemia. Western blot analysis revealed an accumulation of the precursor protein of CPE in the ischemic cortex in vivo and in ischemic cortical neurons in vitro. Detailed metabolic labeling experiments on ischemic cortical neurons showed that ischemic stress caused a blockade in the proteolytic processing of CPE. When mice lacking an active CPE protease were subjected to a sublethal episode of focal cerebral ischemia, abundant TUNEL-positive cells were seen in the ischemic cortex whereas only a few were seen in the cortex of wild-type animals. These findings suggest that ischemia has an adverse impact on the neuropeptide-processing system in the brain and that the lack of an active neuropeptide-processing enzyme exacerbates ischemic brain injury.
Neuropeptides play important roles in regulating the brain's response to various stimuli and stresses, including ischemia. After brain ischemia, expression levels of a number of neuropeptides are upregulated. Examples include nerve growth factor, brain-derived neurotrophic factor, dynorphin, enkephalin, neuropeptide Y, and chromogranins (Akins et al., 1996; Allen et al., 1995; Heron et al., 1996; Hokfelt et al., 2000; Johnson et al., 1994; Marti et al., 2001). Ablating certain neuropeptides or blocking their action in the brain exacerbates ischemic injury, whereas providing such neuropeptides reduces ischemic injury (Baskin et al., 1994; Cheung and Cechetto, 2000; Endres et al., 2000; Schabitz et al., 2000). Thus, an ischemia-induced increase in the expression of certain neuropeptides is considered a protective mechanism of the brain against ischemic stress.
In neuronal and endocrine cells, virtually all known neuropeptides and peptide hormones are processed from large precursor molecules by the action of a set of processing enzymes in the secretory pathway. Such processing enzymes include, but are not limited to, members of the proprotein convertase (PC endoprotease) family, several carboxypeptidases, and an amidating enzyme (Seidah and Prat, 2002; Steiner, 1998; Taylor et al., 2003; Wei et al., 2003; Zhou et al., 1999). Studies conducted in recent years have shown that expression levels of these processing enzymes are highly responsive to stresses such as seizure (hippocampus), electroconvulsion (hippocampus), irradiation (whole brain), mechanical injury (sciatic nerves), and light/dark switch (retina) (Bhat et al., 1993; Marcinkiewicz et al., 1997; Marcinkiewicz et al., 1998; Meyer et al., 1996; Noel et al., 1998; Schlamp and Nickells, 1996). These observations suggest an essential role of neuropeptide processing in the stress response of the nervous system. In a previous study on ischemic rat brains that used a subtractive library screening approach, we identified carboxypeptidase E (CPE) (Jin et al., 2001). A transient increase in expression levels of mRNA and the protein for CPE was observed in the hippocampus after global ischemia, suggesting a possible involvement of neuropeptide-processing enzymes in the brain's response to ischemia. An important question that was not addressed by the previous study, however, is whether increased expression of CPE after brain ischemia will result in an increase in the level of a properly processed CPE protease. In a recent study, Lee et al. (2001) showed that neurotrophins and unprocessed proneurotrophins have opposing effects on cell survival (pro-cell survival vs. pro-cell death, respectively). In this regard, the ability of brain cells to process neuropeptides properly may compose a critical aspect of the brain's response to ischemic insults.
Carboxypeptidase E is one of the key neuropeptide-processing enzymes in the brain. It removes C-terminal basic amino acid residuals after they are exposed by the action of endoproteolytic processing enzymes (e.g., PCs). Neuropeptide processing mediated by CPE and PCs is a regulated process that is mechanistically different from protein degradation. A role of CPE in neuropeptide sorting in the regulated secretory pathway has also been proposed (Cool et al., 1997). CPE is initially synthesized as a proenzyme. The proteolytic cleavage of its propeptide and the function of CPE are regulated by pH and calcium concentration in the regulated secretory pathway, with the pH playing a more predominant role (Fricker and Devi, 1993; Nalamachu et al., 1994; Song and Fricker, 1995a, 1995b). When intraluminal balances of pH or calcium concentration are disturbed (e.g., by drug treatment), the proteolytic processing of CPE can be perturbed; in parallel, the processing of PC proteases that work in concert with CPE can also be attenuated (Guest et al., 1997; Song and Fricker, 1995a). Such failures in the biosynthesis of processing enzymes are accompanied by a blockade in neuropeptide processing (Guest et al., 1997; Song and Fricker, 1995a). Hence, a neuropeptide-processing system involving CPE is subject to change when cellular conditions change. The pathology of brain ischemia includes a rapid influx of calcium from extra-cellular space and an efflux of calcium from the endoplasmic reticulum, accompanied by tissue acidosis (Dirnagl et al., 1999; Nedergaard et al., 1991). The consequences of these changes have been described in great detail for mechanisms of ischemia-induced, delayed cell death in brain ischemia. Little is known about how ischemia-induced cellular changes may affect neuropeptide-processing enzymes. In this study, we investigated several aspects of CPE concerning its biosynthesis after ischemic stress, using both in vivo and in vitro ischemia models. We also investigated how the availability of an active CPE might determine the extent of ischemic brain injury by studying CPE-deficient animals. Our results show that brain ischemia attenuates the proteolytic processing of CPE in cortical neurons and that animals lacking an active CPE are more prone to ischemic brain injury.
MATERIALS AND METHODS
Transient focal cerebral ischemia
All animal experiments were performed in accordance with the National Institutes of Health's Guide for the Care and Use of Laboratory Animals. The Institutional Animal Care and Use Committee of Legacy Research approved the protocols for inducing transient focal cerebral ischemia with the suture method in rats (Longa et al., 1989; Shimizu et al., 2001) and mice (Barone et al., 1993; Stagliano et al., 1999).
Briefly, adult (250–300 g) male Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA, U.S.A.) were anesthetized with 4% isoflurane in 70% nitrous oxide (N2O) and 30% oxygen (O2), maintained with 1% isoflurane in 70% N2O/30% O2, and endotracheally incubated and mechanically ventilated by a rodent ventilator (Harvard Apparatus, Holliston, MA, U.S.A.). Rectal temperature was maintained at 37.0° ± 0.5°C with a heating pad (Harvard Apparatus). Brain temperature was measured using a temporalis muscle-implanted thermocouple (Omega Engineering, Inc., Stamford, CT, U.S.A.) and also controlled within the range 37.5° ± 0.5°C by a heating blanket. Under an operating microscope, the bifurcation of the common carotid artery was exposed and the external carotid artery ligated. The internal carotid artery was isolated, its extracranial branch was ligated, and a 3–0 silk suture was introduced into the lumen of the internal carotid artery and advanced 20 to 22 mm to occlude the middle cerebral artery. Reperfusion was achieved by the withdrawal of the suture. Sham operated animals (control) underwent the same surgical procedure but the suture was not advanced to the middle cerebral artery. At the end of reperfusion, animals were killed under anesthesia and brain tissues were processed accordingly for different analyses (described later). For mice, ischemia was also induced by middle cerebral artery occlusion (MCAO). Male Cpefat/fat mice (BKS.HRS-Cpefat/J) (25–30 g, approximately 10 weeks old) and their wild-type littermates were purchased from the Jackson Laboratory (Bar Harbor, ME, U.S.A.). This mouse strain is of the C57BL/J background, thus allowing us to adapt MCAO procedures established on wild-type C57BL/J mice without modification. Under anesthesia with 4% isoflurane in 70% N2O/30% O2, MCAO was performed with a 7–0 mono-filament nylon surgical suture, which was introduced into the internal carotid artery lumen and advanced 7 or 8 mm past the common carotid artery bifurcation. Reperfusion was achieved by the withdrawal of the suture. At the end of reperfusion, animals were sacrificed under anesthetization. Frozen coronal brain sections (12-μm thick) were cut for cell death analyses.
For both rats and mice, relative regional cerebral blood flow (rCBF) was measured by laser-Doppler flowmetry (Transonic Systems Inc., Ithaca, NY, U.S.A.).
Cell cultures and in vitro simulated ischemia
Primary mouse cortical neurons or cortical astrocytes were cultured according to protocols previously described by this laboratory (Xiong et al., 1999; Zhao and Flavin, 2000). Briefly, time-pregnant mice (E18; Charles River Laboratory) were anesthetized with halothane followed by cervical dislocation. Fetuses were rapidly removed and placed in cold Hanks’ solution. The cerebral cortices from 10 to 12 embryos were dissected and incubated with 0.05% ethylenediaminetetraacetic edetic acid in phosphate-buffered saline (PBS) for 10 minutes at 37°C, followed by trituration with flame-polished glass pipettes. For neuronal cultures, dispersed cortical cells were seeded onto poly-L-ornithine-coated 35-mm dishes at a density of 1 × 106 cells/dish. Cells were maintained in Eagle's minimum essential medium supplemented with 10% horse serum (Invitrogen, Carlsbad, CA, U.S.A.) at 37°C in a humidified atmosphere equilibrated with 5% CO2. Five-fluoro-2-deoxyuridine (5 μmol/L) and uridine (5 μmol/L) were added 72 hours after seeding to suppress the growth of glial cells. Cells were used for experiments 12 to 14 days after seeding. For astrocyte cultures, cortical cells were seeded in 75-cm2 flasks at a density of 6 × 105 cells/cm2 in Eagle's minimum essential medium supplemented with 10% fetal calf serum. The culture medium was changed 24 hours after plating and every 3 or 4 days thereafter. On day 12, the flask containing the astrocyte culture was shaken on a rotatory shaker at 800 rev./min for 1 hour. Suspended cells were discarded, and the remaining adherent monolayer of astrocytes was dissociated by trypsin and reseeded onto 35-mm dishes at a density of 1 × 106 cells/dish. Simulated ischemia was induced in cultured neurons or astrocytes by oxygen and glucose deprivation (OGD) (Goldberg and Choi, 1993). For OGD, cells were incubated in glucose-free, serum-free, and glutamine-free DMEM (Invitrogen) in an anaerobic chamber (Forma Scientific, Marietta, OH, U.S.A.) equilibrated with 85% N2/5% CO2/10% H2 for desired periods.
In situ hybridization
Plasmid pGEM.7Zf-CPE (gift of Dr. Lloyd D. Fricker at Albert Einstein College of Medicine, Bronx, NY, U.S.A.) carrying cDNA for rat CPE was digested with restriction enzyme Hind III or Eco RI (Promega, Madison, WI, U.S.A.) to generate linearized templates for antisense or sense (control) cRNA probes, respectively. A 1-kb digoxigenin (DIG)-UTP-labeled antisense or sense probe was prepared using T7 (antisense) or SP6 (sense) polymerase, respectively, with a commercial RNA Labeling Kit (Roche Diagnostics, Indianapolis, IN, U.S.A.) following the manufacturer's instruction.
Paraffin-embedded coronal sections of rat brains were cut at 6-μm thickness and placed onto Superfrost glass slides (Fisher Scientific Ltd, Loughborough, U.K.). Hybridization was performed using protocols previously described (Chen et al., 2001; Jin et al., 2001), with a modification in the detection method. Briefly, sections were dewaxed by xylene and rehydrated through a descending alcohol gradient, washed with PBS, and then treated with 20-μg/mL Proteinase K (Sigma, St. Louis, MO, U.S.A.) in PBS for 10 minutes at room temperature. After washes with PBS, sections were incubated with a prehybridization buffer (50% formamide, 5X SSC, 1X Denhardt's solution, 5% dextran sulfate, 20-mmol/L dithiothreitol, 0.1% sodium dodecyl sulphate (SDS), 250-μg/mL yeast tRNA, 500-μg/mL salmon sperm DNA) for 1 hour before hybridization with DIG-cRNA probes (2 μg/mL) in the same buffer. Hybridization was performed at 42°C overnight in a sealed container equilibrated with 50% formamide. The next day, sections were washed twice with 2X SSC and treated with RNase A (30 μg/mL; Roche) in PBS at 37°C for 30 minutes followed by two washes with 2X SSC at room temperature and 1 wash with 1X SSC at 54°C. After another wash with PBS, sections were subjected to immunocytochemical analysis of DIG (described in the following section).
Fluorescent immunocytochemistry
Single- or double-labeling immunocytochemistry was performed using previously described protocols (Jin et al., 2001). Unless indicated otherwise, paraffin-embedded brain sections were used. For immunocytochemistry, sections were dewaxed by xylene and rehydrated through a descending alcohol gradient. After incubation with a blocking solution (2% horse serum, 0.1% bovine serum albumin in PBS) to reduce nonspecific bindings, sections were incubated with an appropriate primary antibody diluted in the blocking solution at 4°C overnight. The next day, after several washes with PBS, sections were incubated with either fluorescein isothiocyanate (FITC)- or Cy3-conjugated secondary antibody (Vector Co., Burlingame, CA, U.S.A.) at a dilution of 1:400, as specified in the figure legends (see Results). Sections were mounted with 4′,6-diamidino-2-phenylindole (DAPI)-containing mounting fluid (Vector Co.) to counterstain nuclei and examined with an epifluorescence microscope (Leica Microsystems, Inc. Bannockburn, IL, U.S.A.) attached with a Magnifire digital color camera (ChipCoolers, Warwick, RI, U.S.A.). The specificity of primary antibodies was verified by incubation without the primary antibody. For double-labeling fluorescent immunocytochemical analysis of DIG and CPE, sections that underwent in situ hybridization procedures were first incubated with a polyclonal rabbit anti-DIG antibody (Roche) followed by incubation with FITC-conjugated goat-anti-rabbit antibody. Then sections were incubated with a monoclonal mouse anti-CPE antibody (BD Biosciences Clontech, Palo Alto, CA, U.S.A.) followed by incubation with Cy3-conjugated goat-anti-mouse antibody. The double-labeled sections were mounted with DAPI-containing mounting solution and examined with an epifluorescence microscope.
Western blot analysis
Western blot analysis of proteins extracted from brain tissues or cultured cells followed standard protocols (Furuta et al., 1997; Jin et al., 2001). Cortices were homogenized into a buffer consisting of 50-mmol/L Tris-HCl, pH 7.5, 150-mmol/L NaCl, 1% Triton X-100, 2-mmol/L edetic acid, and a cocktail of protease inhibitors. For cultured cells, cells were trypsinized and resuspended with the same buffer used in brain homogenization. Debris and insolubles in brain homogenates and cell extracts were removed by centrifugation at 10,000 g for 10 minutes at 4°C after three cycles of freezing/thawing of the sample. Protein concentrations of cleared supernatants were determined by the Bradford method (Sigma). Fifty micrograms of protein were fractionated by SDS polyacrylamide gel electrophoresis (10% slab gel), blotted onto an Immobilon-P membrane (Millipore, Billerica, MA, U.S.A.), probed with an appropriate primary antibody (specified in the figure legends) followed by detection with the enzyme-catalyzed chemiluminescence method (NEN Life Science Products, Boston, MA, U.S.A.). After enzyme-catalyzed chemiluminescence analysis, blots were stained with Coomassie Blue to verify the evenness of protein loadings among different samples to be compared.
Metabolic labeling of cultured cells
Metabolic labeling of cultured cortical neurons was performed as described by Zhou et al. (1998). Cells were plated onto four-well plates coated with poly-L-ornithine. For labeling, two wells of cells of each treatment group (control, 30-minute OGD, 60-minute OGD, and 60-minute OGD with 24 hours of recovery) were rinsed with methionine-deficient DMEM (Invitrogen) and pulse-labeled for 1 hour with [35S]methionine (Amersham Life Science, Piscataway, NJ, U.S.A.; 1,000 Ci/mmol) suspended in methionine-deficient DMEM (1 mCi/mL, 250 μL/well). After the pulse, cells in one well were extracted immediately (pulse only) whereas cells in the other well were further incubated for another hour in a nonradioactive, complete, serum-free DMEM medium containing 0.1-mg/mL bovine serum albumin (chase incubation). At the end of the chase incubation, chase media were collected and cellular proteins were extracted. The extraction buffer for pulse or chase cells consisted of 50-mmol/L Tris-HCl pH 7.4, 150-mmol/L NaCl, 0.5% NP-40, 1% TX-100, 0.1% sodium deoxycholate, 0.1% SDS, 2-mmol/L edetic acid, and a cocktail of protease inhibitors. Insolubles were removed by centrifugation at 10,000 g for 10 minutes at 4°C. A small aliquot (1%, v/v) of cleared cell extracts from pulse labeling was precipitated with 10% trichloroacetic acid and quantified by scintillation counting to confirm the success of incorporation of [35S]methionine into cellular proteins. CPE proteins in cell extracts and chase media were immunoprecipitated with 4 μL monoclonal mouse anti-CPE antibody (BD Biosciences Clontech) and 20 μL immobilized Protein-A (Pierce, Rockford, IL, U.S.A.) in the same buffer used for cellular protein extraction for Western blotting. Immunoprecipitated CPE proteins were fractionated by 10% SDS polyacrylamide gel electrophoresis. Gels were fixed with 10% acetic acid/20% ethanol for 20 minutes, soaked with the Amplify solution (Amersham), then dried and exposed to x-ray film.
Cell death analysis
Cresyl violet staining (Nissl staining) and immunocytochemistry of NeuN (Chemicon, Temecula, CA, U.S.A.), a neuron marker, were performed on frozen brain sections of Cpefat/fat mice under normal conditions to evaluate any possible pathohistologic abnormalities of the cortex in this mouse strain. Ischemia-injured cells in brain sections of Cpefat/fat mice and their wild-type littermates were identified by detecting DNA fragmentation using the terminal deoxynucleotidyl transferase-mediated 2′-deoxyuridine 5′-triphosphate-biotin nick end labeling (TUNEL) method with a commercial kit (Roche), following the manufacturer's instruction. Labeled sections were mounted with DAPI-containing mounting solution. TUNEL-positive cells were counted in three 20× cortical fields and summed for each brain. The percentages of TUNEL-positive cells were compared between Cpefat/fat mice and their wild-type littermates by t-test, where a value of P ≤ 0.05 was accepted as significant. Frozen sections of ischemic brains were also analyzed by cresyl violet staining to reveal the extent of tissue injury. For primary cultures of cortical neurons and cortical astrocytes, OGD-induced cell death was determined by measuring the lactate dehydrogenase activity in the conditioned medium using a commercial kit (Roche) following the manufacturer's instruction. Lactate dehydrogenase levels in Triton X-100-permeabilized cell cultures were defined as 100%.
RESULTS
Differential changes of levels of mRNA and protein for CPE in ischemic rat brains
Carboxypeptidase E is widely expressed in neuronal cells in different regions of the brain, including the cortex and the striatum (Dong et al., 1999; Jin et al., 2001; Schafer et al., 1993), the two major regions that are injured by MCAO. In this article, we primarily describe the cortical region and cultured cortical cells, using them as our principle targets. In control brains, CPE immunore-activity was detectable in both the soma of neuronlike cells and in fibers in layers II through IV of the cortex. CPE-positive cells were also seen in layers V and VI of the cortex (not shown). The signal for CPE mRNA was readily detectable in the perinuclear region of many cortical cells (Fig. 1A). After 100 minutes of MCAO and 24 hours of reperfusion, CPE immunoreactivity was markedly decreased in the cortex, especially in fibers. Dense CPE immunoreactivity remained detectable in some cortical cells. The signal for CPE mRNA remained strong or even enhanced in many cortical cells in the ischemic brain (Fig. 1A). To further determine if a possible reciprocal relationship may exist between the protein level and the mRNA level for CPE within the same cells after brain ischemia, double-labeling immunocytochemistry and in situ hybridization for CPE were performed on the same brain sections. As illustrated in Fig. 1B, in the control cortex, strong CPE protein immunoreactivity was readily detected in many neuronlike cells, whereas the CPE mRNA signal was relatively weak in the same cells. In the ischemic cortex, strong CPE mRNA signal was detected in cortical cells in which little or weak CPE protein immunoreactivity was seen.
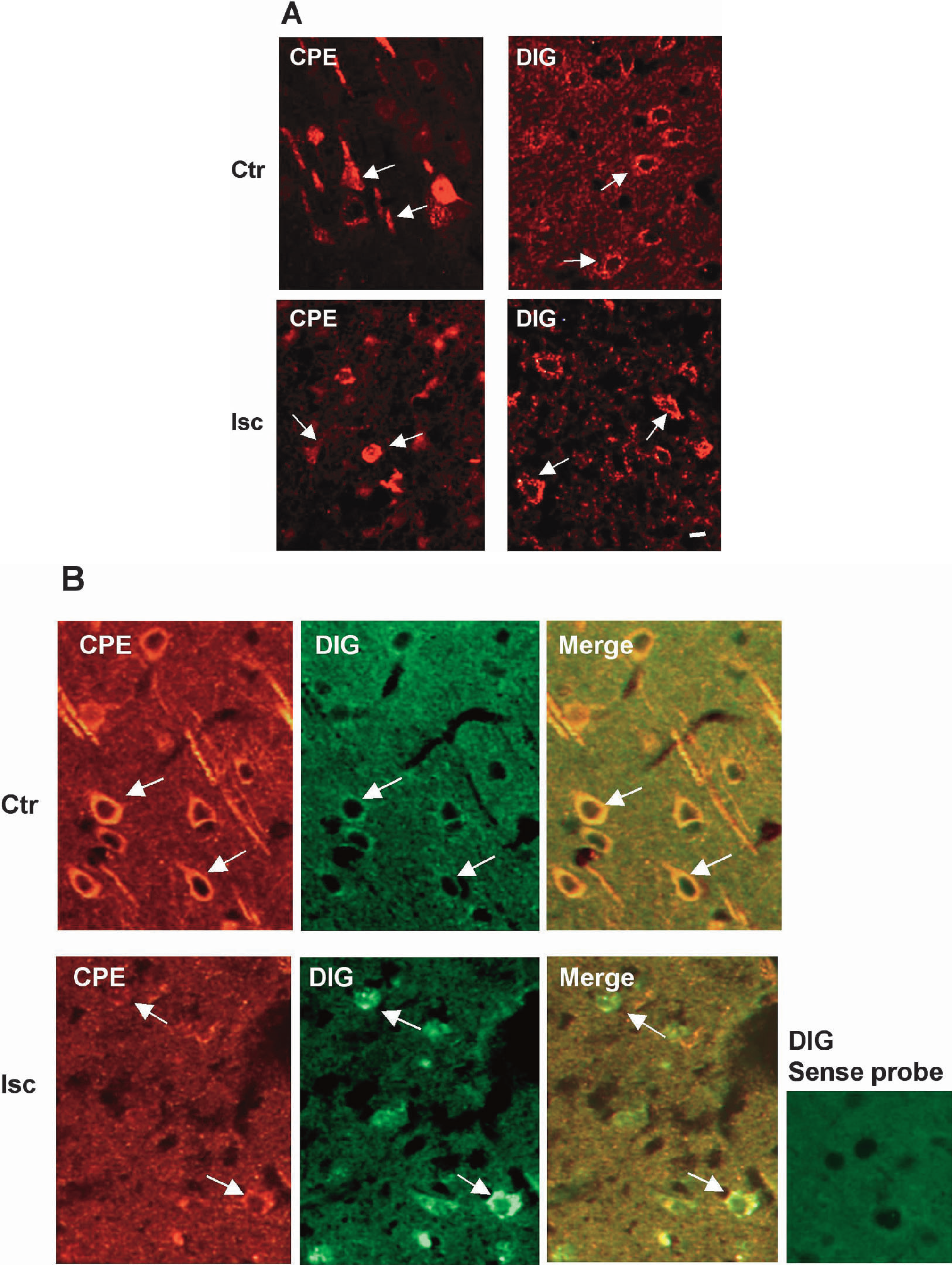
Analyses of CPE expression in the ischemic cortex. Rats were subjected to 100-minute MCAO followed by 24 hours of reperfusion. At the end of reperfusion, animals were perfused with 4% formaldehyde in PBS under anesthesia and paraffin-embedded brain sections were prepared. (
Altered molecular forms of CPE protein in ischemic rat brains
Next, we analyzed CPE protein in the cortex by Western blotting (Fig. 2) to determine its molecular forms and uncover possible changes in the biosynthesis of CPE after brain ischemia. In the control cortex, two major forms of CPE protein, 55 kDa and 53 kDa, were detected, presumably representing the proenzyme form of CPE (pro-CPE) and the cleaved form of CPE, respectively. In the ischemic cortex, the amount of the 53-kDa CPE protein was decreased. The loss of the 53-kDa CPE protein was accompanied by the appearance of a 54-kDa CPE protein and a 59-kDa CPE protein. The presence of the 59-kDa CPE protein was most evident at the 72-hour reperfusion time point. The identities of the 54-kDa and the 59-kDa CPE proteins are not known; they presumably represent pro-CPE or CPE proteins with different glycosylation status.
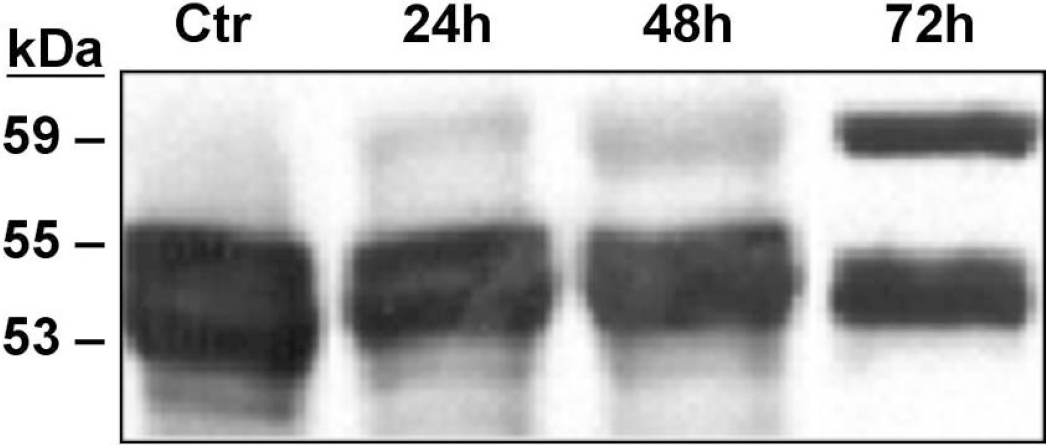
Western blot analysis of CPE protein in ischemic cortices. Rats were subjected to 100-minute MCAO followed by 24, 48, and 72 hours reperfusions. Protein was extracted from control (Ctr) and reperfused brains, fractionated by SDS polyacrylamide gel electrophoresis and blotted onto a polyvinyl difluoride membrane. The membrane was probed with a monoclonal mouse anti-CPE antibody (1:2,000; the same antibody used for immunocytochemistry in Fig. 1) that recognizes both pro-CPE and CPE proteins (antigen: amino acids 49–200 of human CPE) and CPE-related protein bands were visualized with the enzyme-catalyzed chemiluminescence method. Comparable results were obtained in independent analyses of two or three brains for each reperfusion time point.
Altered biosynthetic processing of CPE protein in ischemic cortical neurons
The previously mentioned analyses of CPE mRNA and protein in ischemic cortices indicate the possibility that brain ischemia altered not only the expression of CPE, but also its biosynthesis. These analyses, however, could not reveal in which cell population the change in molecular forms of CPE took place after ischemia. The expression of CPE in cultured astrocytes has been reported (Klein and Fricker, 1992). Although we have not detected the expression of CPE protein at an appreciable level in cortical astrocytes of adult rat brains by immunocytochemical means (Jin et al., 2001), this is probably due to the relatively poor ability of glial cells in storing secretory proteins. We analyzed CPE proteins in purified primary cultures of cortical neurons and cortical astrocytes by Western blotting. In our experimental settings, 60-minute simulated ischemia (OGD) caused little damage to cortical astrocytes but substantial delayed cell death in cortical neurons. Death rates (lactate dehydrogenase release) of 3.7 ± 0.2% and 45.9 ± 4.8% (mean ± SD; four independent cultures) were observed with ischemic (OGD-treated) cortical neurons when analyzed 1 hour and 24 hours after the OGD, respectively. As shown in Fig. 3, 60-minute OGD resulted in an accumulation of pro-CPE-sized protein in cortical neurons (Fig. 3A), but not in cortical astrocytes (Fig. 3B). There was no apparent change in total cellular content of CPE proteins in ischemic cells. When ischemic cortical neurons were further analyzed by Western blotting using a pro-CPE-specific antibody (Varlamov et al., 1997), it was evident that the accumulated high molecular weight CPE protein in ischemic neurons was pro-CPE (Fig. 3C). The accumulation of pro-CPE in ischemic neurons in vivo was demonstrated by the presence of pro-CPE-positive, neuron like cells in the ischemic cortex; such cells were not seen in the control cortex (Fig. 3D).
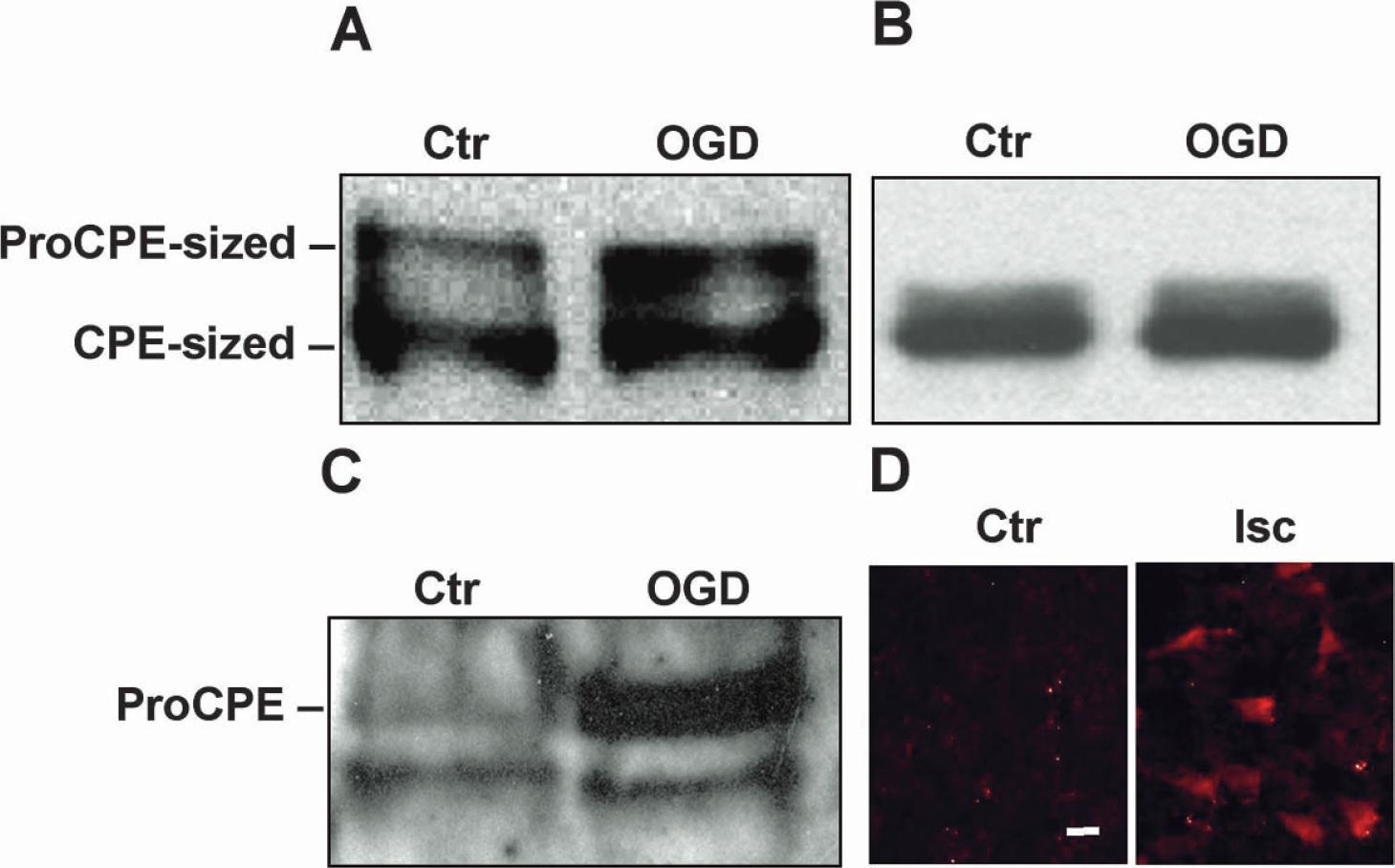
Accumulation of pro-CPE protein in cultured ischemic cortical neurons but not in ischemic astrocytes. Primary cultures of embryonic cortical neurons and astrocytes were subjected to 1-hour OGD treatment followed by 24 hours of recovery incubation in the regular growth medium. At the end of the recovery incubation, cellular proteins from the control (Ctr) and OGD-treated (OGD) cells were extracted and analyzed by Western blotting. (
To investigate molecular mechanisms for the accumulation of pro-CPE in cortical neurons after ischemia, or to see if ischemic stress would attenuate the processing of newly synthesized CPE protein, pulse-chase metabolic labeling analysis was performed on ischemic cortical neurons. Cells were labeled with [35S]methionine after OGD with or without a 24-hour recovery incubation under normal cultural conditions. Isotope-labeled CPE proteins were extracted from labeled cells and conditioned media, immunoprecipitated with a monoclonal mouse anti-CPE antibody and analyzed by SDS polyacrylamide gel electrophoresis and autoradiography. The results (Fig. 4) show that under normal conditions, the appearance of the 53-kDa CPE protein could be detected during the 1-hour pulse-labeling period. When cells were labeled immediately after 30-minute OGD, little 53-kDa CPE protein was produced during the pulse-labeling period, though it could be detected during the subsequent chase incubation. When the OGD period was extended to 60 minutes, the production of the 53-kDa CPE was not detectable either during the pulse-labeling period or during the chase incubation. There was no substantial production of the 53-kDa protein in cells that underwent 60-minute OGD and 24 hours of recovery. Hence, OGD treatment attenuated the proteolytic processing of CPE in cortical neurons in an OGD dose-dependent manner. The blockade took place before the occurrence of substantial cell death and was irreversible after a prolonged OGD.
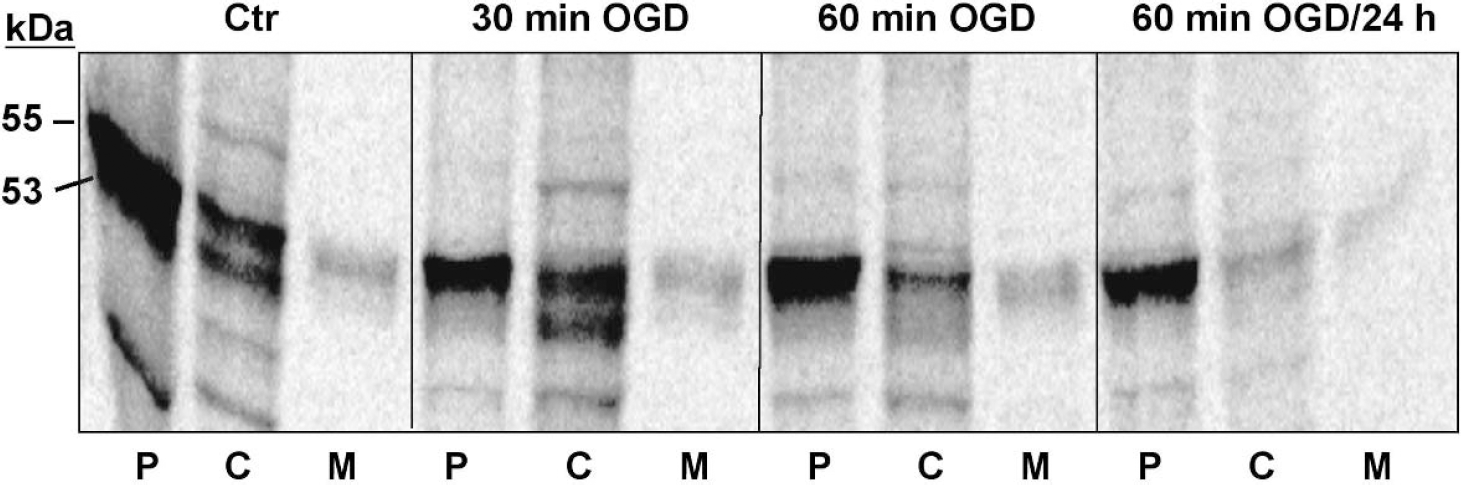
Metabolic labeling analysis of CPE biosynthesis in ischemic cortical neurons. Cultured cortical neurons with or without OGD treatment were labeled for 1 hour with [35S]methionine followed by 1-hour chase incubation in a complete, nonradioactive medium as described in Materials and Methods. At the end of pulse labeling or chase incubation, cellular proteins were extracted and chase media were collected. CPE proteins in pulse-labeled (P) and chase-incubated (C) cell extracts and chase media (M) were isolated by immunoprecipitation with a monoclonal mouse anti-CPE antibody and immobilized Protein-A. The immune complexes were then analyzed by SDS polyacrylamide gel electrophoresis and autoradiography. Similar changes in CPE processing after OGD were seen in another independent experiment. Ctr, control; 30 min OGD and 60 min OGD, cells were labeled immediately after 30-minute OGD and 60-minute OGD, respectively; 60 min OGD/24 h, cells were labeled after 60-minute OGD and 24 hours of recovery.
Animals lacking an active CPE protease are more prone to ischemic brain injury
Cpefat/fat mice are a mouse strain with a spontaneous mutation in the CPE gene that results in the production of an inactive CPE protein (Fricker et al., 1996; Naggert et al., 1995), providing an excellent animal model to study how the lack of an active CPE influences the brain's response to ischemia. First, we analyzed the brain of Cpefat/fat mice by cresyl violet staining and immunocytochemistry of NeuN, a neuronal cell marker. Under normal conditions, no noticeable cell injury and abnormality in the cortical morphology were seen in the cortex of Cpefat/fat brains (Fig. 5A). The density of neuronal cell population in the cortex of Cpefat/fat brains appeared to be comparable with that of wild-type brains (Fig. 5B). When Cpefat/fat mice were subjected to MCAO, the rCBF levels responded to MCAO procedures accordingly. The suture insertion reduced rCBF levels to 24.5 ± 12.72% and 27.15 ± 7.32% of pre-MCAO levels in wild-type and Cpefat/fat mice, respectively (n = 5 each group; P = 0.384). Interestingly, significantly more TUNEL-positive cells were seen in the cortex of Cpefat/fat mice than in the cortex of wild-type mice after 30-minute MCAO (Figs. 6B and 6C). Furthermore, substantial losses of Nissl staining, an indication of infarction, were found in both the cortex and the striatum of ischemic Cpefat/fat brains whereas such a loss was restricted to the striatum in ischemic wild-type brains (FIG. 6D). These results strongly suggest that CPE plays a critical role in brain cell survival after ischemic stress.
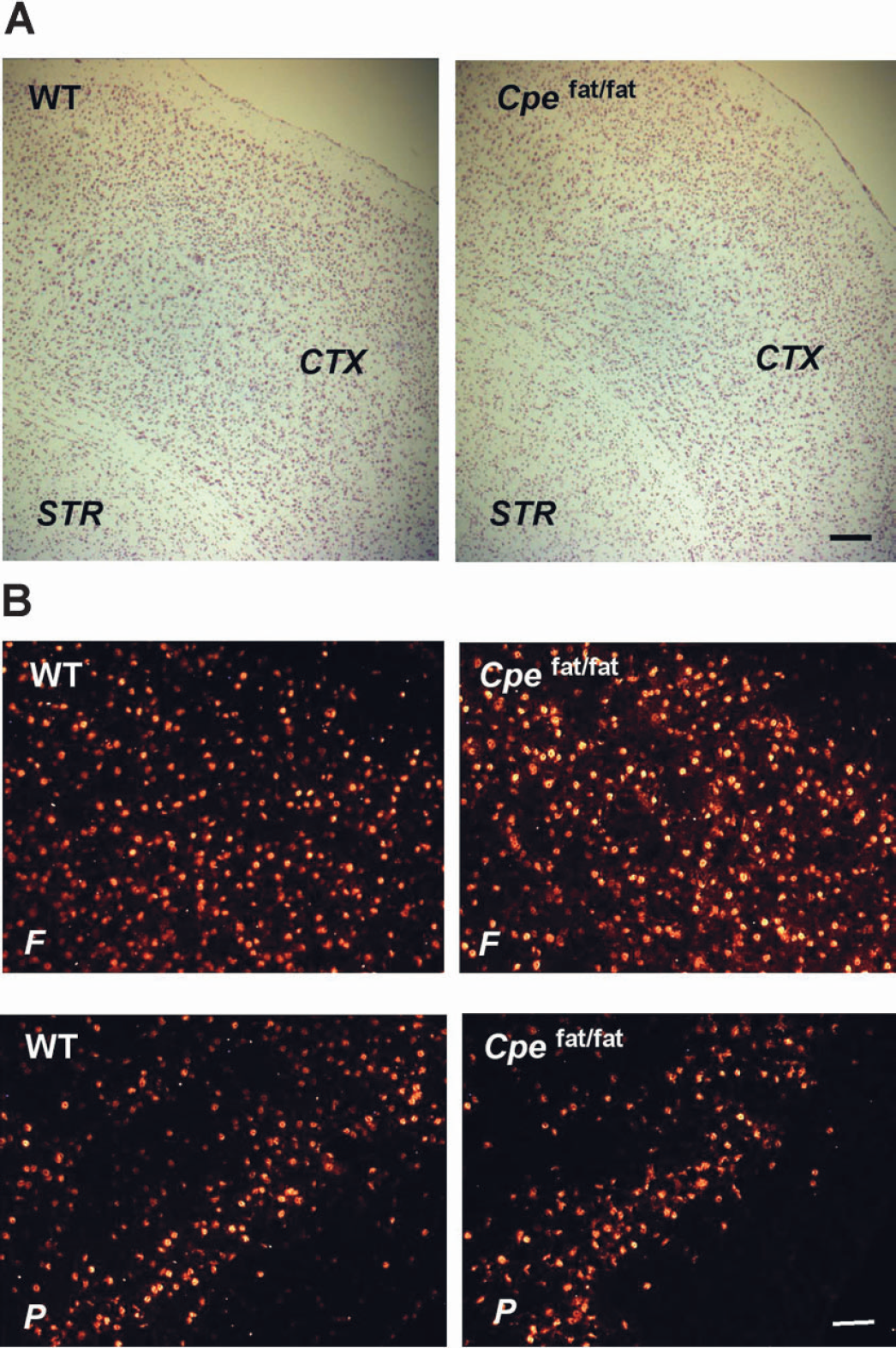
Examination of the cortex of Cpefat/fat mice. Frozen brain sections from Cpefat/fat mice (Cpefat/fat) and their wild-type litter-mates (WT) were prepared under normal conditions (no surgical procedures). Sections were analyzed by cresyl violet staining (
Analyses of ischemic brain injury in Cpefat/fat mice. Frozen brain sections were prepared from Cpefat/fat mice (Cpefat/fat) (n = 3) and their wild-type littermates (WT) (n = 3) after 30-minute MCAO and 24 hours of reperfusion. No TUNEL-positive cells were seen in the contralateral, non-ischemic cortex (
DISCUSSION
The major questions we asked in this study were how brain ischemia may affect the neuropeptide processing system, an essential cellular component of brain cells in producing signaling neuropeptides, and how the lack of a neuropeptide processing enzyme may affect the brain's response to ischemic stress. Our results on CPE in the ischemic cortex and cultured cortical neurons showed that brain ischemia not only altered the expression levels of CPE, but also attenuated its proteolytic processing. Moreover, animals lacking an active CPE protease had exacerbated brain injury after a brief focal ischemia.
Using semi-quantitative neuroanatomical means, we identified reciprocal changes of CPE mRNA and protein in the same cells in the ischemic brain (FIG. 1) when examined at 24 hours reperfusion. It is known that prolonged ischemia will result in an inhibition of protein synthesis and/or RNA export from nuclei, whereas gene transcription activities may remain high for an extended period after an ischemia episode. Ischemic stress can also lead to protein misfolding and instability. These established mechanisms may explain the aforementioned changes in expression levels of CPE in the ischemic brain. As introduced earlier, CPE and PC processing enzymes are pH- and calcium-regulated proteases. Intracellular pH and calcium concentrations are known to change in ischemic cells. Nitric oxide, a candidate mediator of ischemic cell death, can reduce CPE activity in vitro (Devi et al., 1994). Our further analyses of CPE proteins in ischemic brains and purified cortical neurons revealed an accumulation of CPE protein in its precursor form and a loss of its processed form. This was shown by the results of several different analyses (Western blotting and immunocytochemistry using antibodies that either recognize both the pro and the processed forms of CPE, or the pro form only) (Figs. 2, 3 and 4). Metabolic labeling analysis of ischemic cortical neurons demonstrated a blockade in the conversion of pro-CPE to CPE (Fig. 4), suggesting a possible mechanism for the change in molecular forms of CPE proteins in ischemic brains and neurons. The consequence of ischemia-induced attenuation in CPE processing remains to be fully elucidated. The conversion of pro-CPE to CPE takes place in post-Golgi secretory vesicles in the regulated secretory pathway of neuronal and endocrine cells (Song and Fricker, 1995a). Thus, attenuation in this processing step is indicative of adverse changes in neuropeptide processing system. Interestingly, the blockade in CPE processing in ischemic neurons took place before the cell death (i.e., changes in CPE processing were detected immediately after the OGD when no substantial lactate dehydrogenase release had occurred). This raises the possibility that neuropeptide processing after an ischemia episode may play a role in the post-ischemia pathology of the brain.
Although ischemia-challenged expression of neuro-peptides has been described in many studies, the exact molecular forms of neuropeptides in ischemic brains remain to be determined. An accumulation of basic amino acid-extended peptides would presumably reflect the loss of CPE activity as seen in Cpefat/fat mice (Che and Fricker, 2002; Che et al., 2001). An early study by Parkinson (1990), however, showed that pro-CPE protein purified from bovine pituitary is enzymatically active when analyzed using synthetic substrate in vitro. At the same time, in addition to changes in CPE after ischemia, there may be adverse changes in endoproteolytic processing activities in ischemic cells, as indicated by our parallel analyses on PC proteases in ischemic brains and cortical neurons (Zhou et al., 2001–2003 unpublished data). Such changes would limit the exposure of basic residues within the sequence of a neuropeptide. In this respect, ischemia-induced changes in neuropeptide processing may not be a mere reflection of changes in CPE processing. Nevertheless, profiles of neuropeptides in ischemic brains need to be thoroughly characterized in future studies.
In the brain of Cpefat/fat mice, many neuropeptides are present in incompletely processed forms (Bures et al., 2001; Che and Fricker, 2002; Che et al., 2001). Differential alterations in the opioid receptor activity in the brain of Cpefat/fat mice have recently been reported (Boudarine et al., 2002). There have been no published studies on this mouse strain in a setting of brain ischemia. Our work showed that Cpefat/fat mice exhibited substantial cortical injury not seen in wild-type mice after a brief focal ischemia (FIG. 6). It is noticeable that there were few TUNEL-positive cells in the brain of Cpefat/fat mice under normal conditions. This implies that the survival of brain cells after ischemia requires de novo synthesis and proper processing of neuropeptides. One issue that needs to be considered is, in addition to deficiencies in neuropeptide processing, whether other abnormalities in Cpefat/fat mice may contribute to the exacerbated ischemic injury in this mouse strain. Cpefat/fat mice have elevated ratio of proinsulins to insulins in both the pancreas and the circulation (hyperproinsulinemia) (Naggert et al., 1995). Male Cpefat/fat mice develop hyperglycemia at about 12 weeks of age (Leiter et al., 1999), a condition that is known to worsen the ischemic brain injury. It is most likely that the increased ischemic brain injury in Cpefat/fat mice seen in this study (using 10-week-old male mice) was primarily due to defective neuropeptide processing. Another factor that may affect the outcome of brain ischemia is local blood flow. An appropriate change in the rCBF level after MCAO procedures, as what was observed in Cpefat/fat mice in this study, does not exclude the possibility that the level of absolute CBF in this mutant strain may be different from that of wild-type mice. Also, in the periphery of the ischemic territory where injury could also occur but the blood flow was not monitored, changes in rCBF may be different in Cpefat/fat mice than in wild-type mice after MCAO. A comparison of responses to brain ischemia among different processing-deficient animal models that differ in phenotypes (e.g., PC2-knockout mice are mildly hypoglycemic;Furuta et al., 1997) would provide additional leads concerning the roles of neuropeptide processing in the pathology of brain ischemia.
In summary, our present study shows a sustained expression of the neuropeptide-processing enzyme carboxypeptidase E after focal cerebral ischemia. The expressed CPE protein, however, is not processed properly in ischemic cortical neurons. Animals lacking an active CPE are more prone to ischemic brain injury. In light of the recent study by Lee and colleagues on the pro-cell death effect of unprocessed neurotrophins (Chao and Bothwell, 2002; Lee et al., 2001), our findings in this study implicate a novel mechanism of ischemic brain cell death that is mediated by neuropeptide processing.
Footnotes
Acknowledgements
The authors thank Dr. David Henshall for his insightful discussions and Sue Crawford for administrative assistance.