
Abstract
Hereditary hemorrhagic telangiectasia (HHT), associated with brain arteriovenous malformations, is caused by a loss of function mutation in either the endoglin (HHT1) or activin receptor-like kinase 1 gene (ALK-1, HHT2). Endoglin heterozygous (Eng+/−)mice have been proposed as a disease model. To better understand the role of endoglin in vascular malformation development, we examined the effect of vascular endothelial growth factor (VEGF) hyperstimulation on microvessels in adult endoglin heterozygous (Eng+/−) mice using an adenoviral vector to deliver recombinant human VEGF165 cDNA (AdhVEGF) into basal ganglia. VEGF expression was increased in AdhVEGF mice compared with the AdlacZ and saline group (P < 0.05) and localized to multiple cell types (neurons, astrocytes, endothelial cells, and smooth muscle cells) by double-labeled immunostaining. VEGF overexpression increased microvessel count for up to 4 weeks in both the Eng+/+ and Eng+/+ groups (Eng+/+ 185 ± 14 vs. Eng+/− 201 ± 10 microvessels/mm2). Confocal microscopic examination revealed grossly abnormal microvessels in eight of nine Eng+/− mouse brains compared with zero of nine in Eng+/+ mice (P < 0.05). Abnormal microvessels featured enlargement, clustering, twist, or spirals. VEGF receptor Flk-1 and TGF-β receptor 1 (TβR1) expression were reduced in the Eng+/− mouse brain compared with control.
Excessive VEGF stimulation may play a pivotal role in the initiation and development of brain vessel malformations in states of relative endoglin insufficiency in adulthood. These observations are relevant to our general understanding of the maintenance of vascular integrity.
Hereditary hemorrhagic telangiectasia (HHT), an autosomal dominant vascular malformation, is a heterogeneous disorder in terms of its clinical manifestations. HHT is caused by a loss of function mutation in either the endoglin (HHT 1) or activin receptor-like kinase 1 genes (HHT 2). Of patients with HHT, 10% to 25% develop life-threatening complications, such as pulmonary or brain arteriovenous malformations (BAVM) (van den Driesche et al., 2003). Endoglin (CD105), a TGF-β1 receptor-associated glycoprotein, is expressed on the surface of endothelial cells of capillaries, veins, and arteries in mammals (Gougos and Letarte, 1990). Endoglin binds TGF-β1 and TGF-β3 by associating with the TGF-β II receptor (Cheifetz et al., 1992) and is a regulator of TGF-β action in endothelial cells. Endoglin-deficient mice died at embryonic day 10.5 to 11.5 because of vascular developmental anomalies demonstrating the requirement of endoglin for angiogenesis (Li et al., 1999). Cerebral lesions in patients with HHT are detected only after they are symptomatic, whereas in the mouse model we are able to investigate the development of the small abnormal vessels seen in this study. The phenotype of the endoglin heterozygous mice mimics human HHT disorder, and some of them develop abnormal vascular lesions with increasing age (Bourdeau et al., 2001; Torsney et al., 2003). The endoglin heterozygous mice provide a unique tool to study the genetic role of microvascular disorders, including BAVM (Satomi et al., 2003).
Vascular endothelial growth factor (VEGF) is a secreted endothelial cell mitogen. VEGF binds its receptors on endothelial cells, activating endothelial cells to release proteases, proliferate, and migrate to form new vascular structures. Using adenoviral vector delivery VEGF (AdhVEGF) in the mouse brain, we have successfully developed a focal brain angiogenesis model (Xu et al., 2003). Clinical studies demonstrate that VEGF is greatly increased in hemorrhagic stroke, cerebral ischemia, Alzheimer disease, and human BAVM (Hayashi et al., 2003; Sonstein et al., 1996; Tarkowski et al., 2002; Zhang et al., 2000). However, the nature of VEGF-induced angiogenesis in the normal adult brain is unknown. A fundamental question that remains to be resolved in understanding the pathobiology of BAVM is whether the regional nature of vascular malformations is germline, such as HHT or venous malformations (Vikkula et al., 1996), or sporadic in nature, such as sporadic BAVM (Arteriovenous Malformation Study Group, 1999; Fleetwood and Steinberg, 2002). It is possible that some subclinical event might locally alter the balance of vascular signaling, such as minor trauma or ischemia in a subject with a genetic predispostion to have altered angiogenic responses.
Based upon clinical observations and an experimental brain angiogenesis model, we hypothesized that abnormal brain microvessel malformation may be initiated, developed, or enhanced in adulthood by focal hyperstimulation of angiogenesis, such as ischemia, trauma, inflammation, and tumors. These disorders induce growth factors and cytokine production and release. To test this hypothesis, we induced focal hyperstimulation experimentally by localized delivery of AdhVEGF into the brain of Eng+/− mice. With this approach, we expect to answer the following questions:
Does VEGF overexpression induce focal angiogenesis in the adult Eng+/− mouse brain?
If so, what is the difference of microvascular morphology between Eng+/− and Eng +/+ mice?
What are the possible roles of VEGF and endoglin in the development of angiogenic responses in adulthood?
METHODS
Animal preparation and genotyping
Procedures for the use of laboratory animals were approved by the institutional animal care and use committee. The Eng+/− mice have been previously described (Arthur et al., 2000; Torsney et al., 2003). Wild type (Eng+/+) mice were generated from the same chimeric founder and interbred among themselves. Both wild type and mutant strains are generation N3 in the C57BL/6 background. For routine genotyping, polymerase chain reaction (PCR) contained two primer pairs simultaneously. For the LacZ gene, primers were AGGTGCGGATTGAAAATGG and CGGTCAGACGATTCATTGG; to detect wild type endoglin gene, primers from the region deleted in the mutant allele were used, that is, ACCATCTTGTCCTGAGTAGCG and TGAGCCTGACGGGAAACTG. Annealing temperature was 58°C, and 35 cycles were used for tail DNA. PCR products were resolved on 1.5% agarose gels.
Induction of angiogenesis in the mouse brain
All designed protocols and procedures were reviewed and approved by the University of California Animal Care and Use Committee. Eng+/− or Eng+/+ littermates at 8 to 10 weeks were used for the studies. Mice were anesthetized using ketamine (80 mg/kg) and Xylazine (0.5 g/kg) intraperitoneally. After placement in a stereotaxic apparatus (David Kopf Instruments, Tujunga, CA, U.S.A.), a midline incision was made, and a hole was drilled in the skull leaving the dura intact (0.6 mm anterior to the coronal suture and 2 mm lateral to the sagittal suture). A 33-gauge pipet needle with 10 μL Hamilton syringe was stereotaxically inserted into the striatum to a final depth of 3 mm. A single injection of 2 μL virus suspension (AdhVEGF or AdlacZ) containing 1 × 108 plaque forming units (pfu) was injected at a rate of 0.2 μL/minute. The needle was left in place for 5 minutes and then gradually withdrawn over the course of 15 minutes. The bone window was sealed, and the skin was closed. Animals were killed at 7, 14, 21, and 28 days after AdhVEGF injection.
Immunohistochemistry
Sections were dried for 30 minutes and prefixed in acetone for 15 minutes at −20°C. The sections were incubated with Laszlo's blocking solution (10 mM Trizma, 500 mM NaCl, and 0.05% Tween-20, pH7.6) for 30 minutes in a moisture chamber to block nonspecific binding and then incubated with rabbit anti-VEGF antibody (1:75 dilution, Neomarker, Fremont, CA, U.S.A.) overnight at room temperature plus 2 hours at 37°C. Sequential double fluorescence staining for NeuN, glial fibrillary acidic protein (GFAP), mouse endothelial cell (MEC), and myosin were incubated at 4°C overnight. Biotinylated secondary antibodies were incubated for 1 hour. Finally, sections were incubated with Rhodamine (Texas Red) and fluorescein isothiocyanate (FITC) conjugated Streptavidin for 60 minutes (see Table 1 for detailed antibodies).
Antibodies
Statistical analysis
Parametric data between Eng+/− and Eng+/+ mice was compared with Student's t-test. Nonparametric data were compared using Mann-Whiney U test. All data were presented as mean ± SD. A probability value of less than 5% was considered statistically significant.
RESULTS
Increased expression of VEGF and angiogenesis in AdhVEGF-transduced mice
We examined the VEGF expression in the adult mouse brain after AdhVEGF gene transfer. Dose response studies showed 2 μL of 1 × 108 particles induce highest VEGF levels without visible side effect. Mortality would occur if the dose was greater than 1 × 109 particle levels (data not shown here). VEGF immunostaining showed that little immunoreactivity could be detected in the AdlacZ-transduced and the saline-treated mice. However, extensive VEGF immunoreactivity could be detected in the AdhVEGF-transduced mouse brain, and this positive staining was localized in the AdhVEGF-injected hemisphere (Fig. 1, top panel). Double-labeled fluorescence studies revealed that neurons, astrocytes, endothelial cells, and smooth muscle cells were colocalized with VEGF in cell bodies after AdhVEGF gene transfer (Fig. 1, middle panel). At day 7, VEGF-positive staining was mainly detected in the neurons. At day 14, VEGF positive staining was detected mostly in the astrocytes, and in some endothelial and smooth muscle cells, but not in neurons. At days 21 and 28, many endothelial cells and smooth muscle cells expressed VEGF, whereas astrocytes and neurons exhibited much less positive staining. These results demonstrated that neurons, astrocytes, endothelial cells, and smooth muscle cells are the main sources of VEGF after AdhVEGF transduction. Next, we counted the number of microvessels in the AdhVEGF-, AdlacZ-, and saline-treated mice using fluorescent lectin staining. Compared with normal brain microvasculature, the numbers of microvessels were not significantly different in either the AdlacZ-transduced or saline-treated mice. However, the number of microvessels in the Adh- VEGF-transduced mice was significantly increased after 2 to 4 weeks of AdhVEGF gene transfer (Fig. 1, bottom panel, P < 0.05).
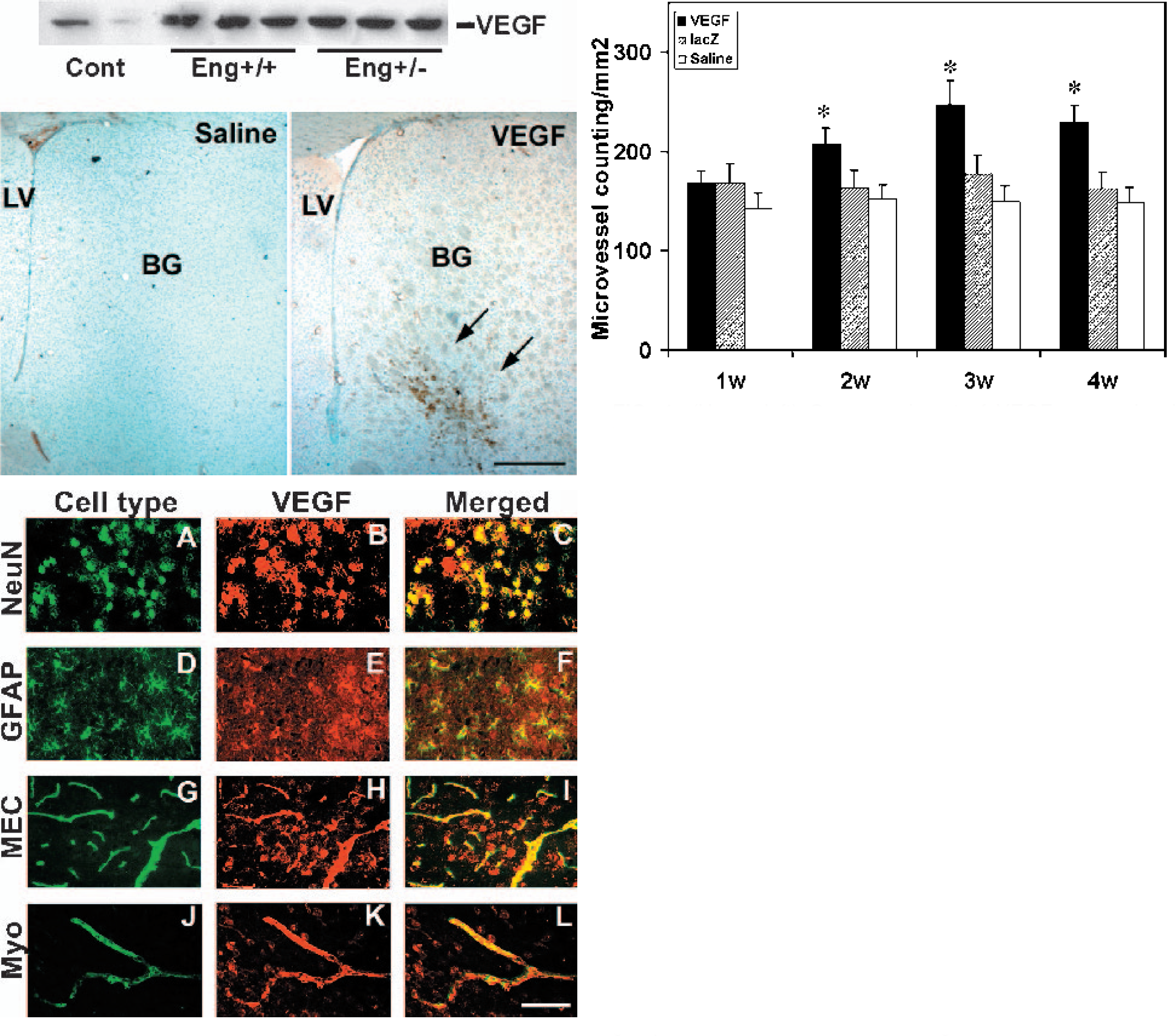
(Upper left) One experiment of VEGF expression in Western blot analysis. VEGF shows a major band at 45 kDa in nonreducing gels. VEGF expression in injection region is greatly increased after 3 weeks of AdhVEGF transduction. VEGF expression is not significantly different between the Eng+/+ and the Eng+/− mice. (Bottom left) VEGF immunostaining was performed after 7 days of AdhVEGF transduction. There is no VEGF positive staining in the saline-injected mice. However, VEGF positive staining was detected in the basal ganglia of AdhVEGF-injected mice (arrows). LV, lateral ventricle; BG, basal ganglia. Bar = 500 μm. (Middle) Photomicrographs show double-labeled immunostaining in the mouse brain after AdhVEGF gene transfer. Green fluorescence staining with anti NeuN, GFAP, MEC, and Myosin antibodies, which identify these positive cells as neurons (A), astrocytes (D), endothelial cells (G), and smooth muscle cells (J). VEGF positive staining (B, E, H, and K, red color) is detected in the ipsilateral hemisphere adjacent to the AdhVEGF-injected region, especially near the needle track. C, F, I, and L show colocalization of green and red color, indicating neurons, astrocytes, endothelial cell, and smooth muscle cells are the source of brain VEGF after AdhVEGF gene transfer. Bar = 50 μm. (Upper right) Bar graph shows the number of lectin-positive staining microvessels in the AdhVEGF-, AdlacZ-transduced, and saline-treated mouse brain after 1 to 4 weeks of AdhVEGF injection. Lectin-positive microvessels are counted at the sections +0.5 and −0.5 mm related to needle track. Values are mean ± SD, N = 6 per group. * P < 0.05, AdhVEGF group vs. AdlacZ or saline treated groups. VEGF, vascular endothelial growth factor.
VEGF increased brain microvessels in both Eng+/− and Eng+/+ mice
Eng+/− and Eng+/+ mice were identified by a standard PCR-based genotyping assay. No visible abnormal behavior or external vascular lesions were detected in the Eng+/− and Eng+/+ mice before experimental intervention at 2 months of age. Neither mortality nor evidence of hemorrhage was observed during the 3 weeks of AdhVEGF gene transfer in either the Eng+/− or Eng+/+ mice.
The first experiment demonstrated that AdhVEGF-induced brain angiogenesis occurred between 2 to 4 weeks. Therefore, we counted the number of microvessels in the Eng+/− and Eng+/+ mice after 3 weeks of AdhVEGF transduction. The number of microvessels in the AdhVEGF injection site was significantly increased in both the Eng+/− and Eng+/+ mice compared with the saline-treated mice (Fig. 2, left panel). However, the increased numbers of microvessels were not significantly different between the Eng+/− and Eng+/+ mice (Fig. 2, right panel, P > 0.05). This shows that AdhVEGF transduction can induce focal angiogenesis in adult mouse brain tissue in both Eng+/− and Eng+/+ mice.
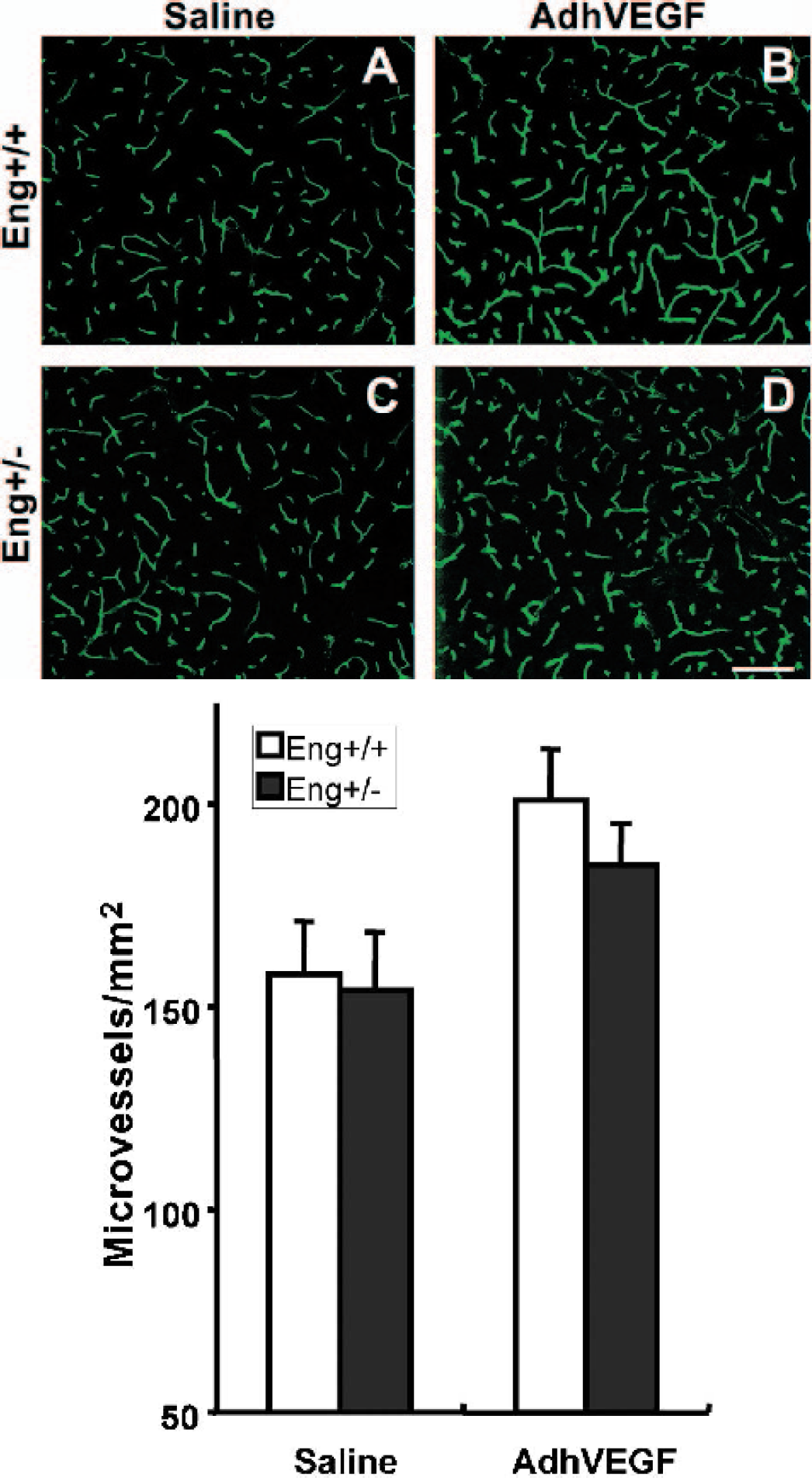
(Upper panel) Lectin-stained microvessels in the Eng+/+ (A and B) and the Eng+/− (C and D) mouse brain after 3 weeks of saline or AdhVEGF gene transduction. The number of microvessels appears comparable in the Eng+/+ and the Eng+/− mouse brain after saline- (A and C) or AdhVEGF-treated (B and D) mice. Bar = 25 μm. (Bottom Bar Graph) Number of lectin staining microvessels in the Eng+/+ and the Eng+/− mouse brain after 3 weeks of saline or AdhVEGF transduction. Values are mean ± SD, N = 9 per group. The number of microvessels was significantly increased after 3 weeks of AdhVEGF transduction in both the Eng+/+ and the Eng+/− mice. However, there was no statistical difference between the Eng+/+ and the Eng+/− mice (P > 0.05).
Abnormal microvessel developed in the Eng+/− mice
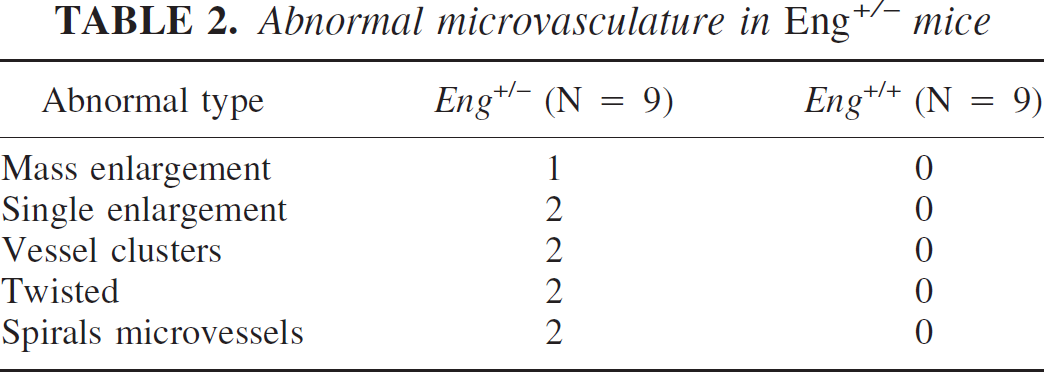
We next examined the microvascular morphologic changes. Forty coronal sections in each animal were examined under confocal microscopy. In the Eng+/+ group, we observed angiogenic changes, such as dilated “mother” vessels and newly formed sprouts, but no abnormal microvasculature was detected in any of the nine mice. However, we observed abnormal microvasculature in eight of the nine Eng+/− mice (Eng+/− vs. Eng+/+; 8/9 vs. 0/9, P < 0.05). These abnormal microvessels usually developed in the ipsilateral cortex or the abundant microvessel areas adjacent to the needle track. These microvessels showed abnormal patterns such as a massive or single enlargement, clustered, twisted, or spiral microvessels (Table 2 and Fig. 3a). In three-dimensional reconstruction sections, these microvessels clearly showed abnormal vascular structure (Fig. 3b). This shows that VEGF hyperstimulation can drive abnormal microvessel formation in the adult Eng+/− mice.
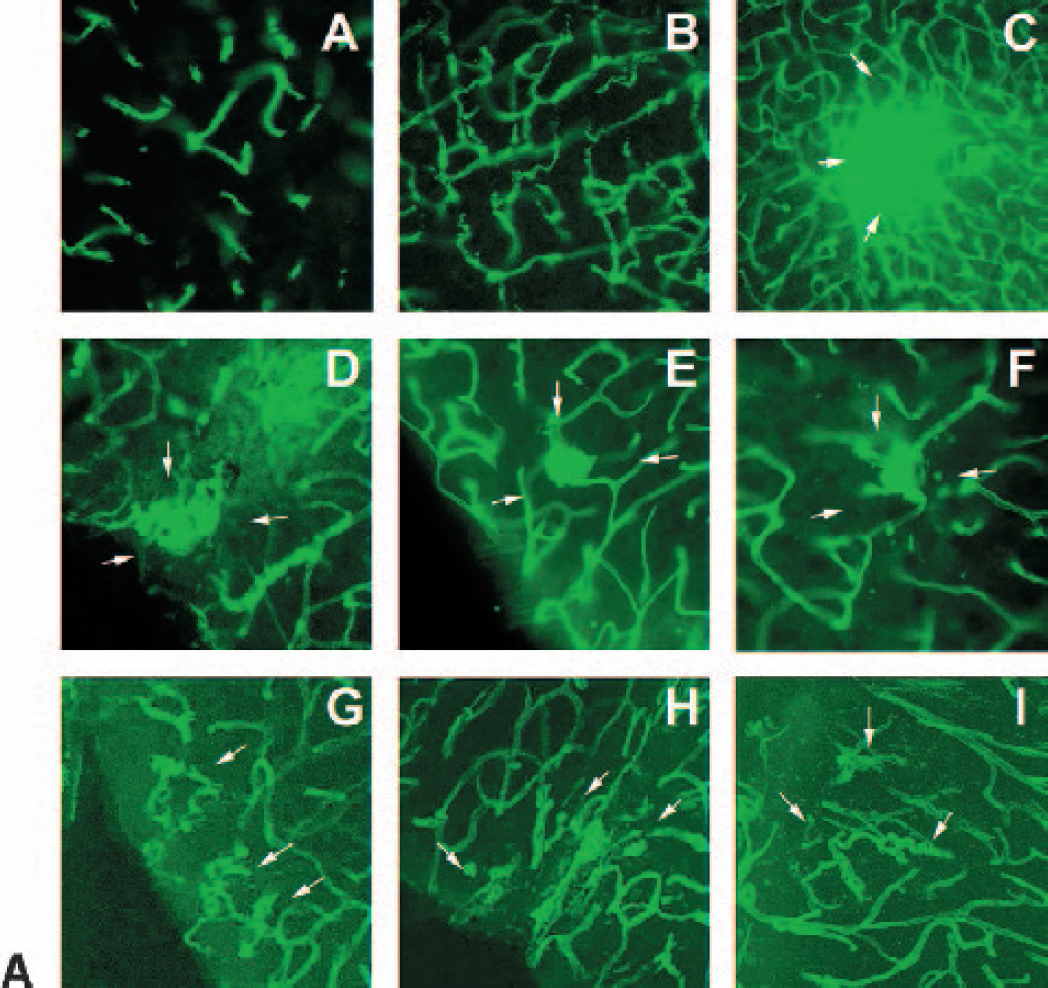
Changes of microvasculature morphology in the Eng+/+ and Eng+/− mouse brain after VEGF overexpression. No abnormal microvasculature after saline injection was detected in the Eng+/− mice (A). Typical angiogenic changes are detected in the Eng+/+ mice after AdhVEGF transduction but no abnormal microvasculature (B). Abnormal microvasculature morphology is detected in the Eng+/− mouse brain. These abnormal microvessels could display a mass (C), single enlargement (D and E), vessel clusters (F), twisted (G and H), or spirals microvessels (I). Arrows indicated the localized abnormal microvasculature. The abnormal microvasculature development is mainly observed in the ipsilateral cortex near the needle track. Bar = 50 μm. VEGF, vascular endothelial growth factor.
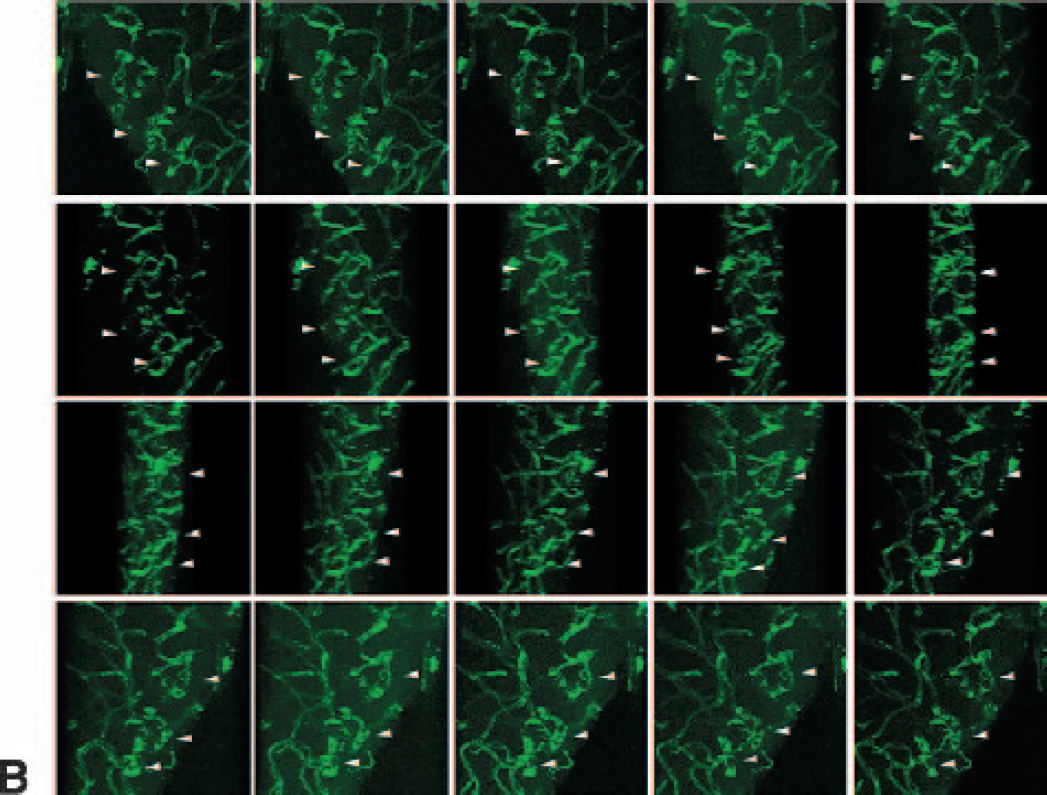
Example of three-dimensional imaging of cerebral microvessels in an Eng+/− mouse using a confocal microscope. A series of 1-μm thick sections are reconstructed, and 20 different angle pictures were taken. The arrows indicate the morphology of abnormal microvessels from different angles. Bar = 50 μm.
Flk-1 expression
To determine the success of AdhVEGF gene transfer, we also measured expression of VEGF and Flk-1 in the Eng+/− mice after AdhVEGF gene transduction. VEGF expression was greatly increased both in the Eng+/− and Eng+/+ mice, and this increase persisted at least 3 weeks after AdhVEGF transduction (Fig. 1, top panel). Flk-1 positive staining was mainly colocalized on the microvessels. Of interest is the fact that the induced level of Flk-1 expression on microvessels in the Eng+/− mice was greatly reduced compared with the Eng+/+ mice (Fig. 4, P < 0.05).
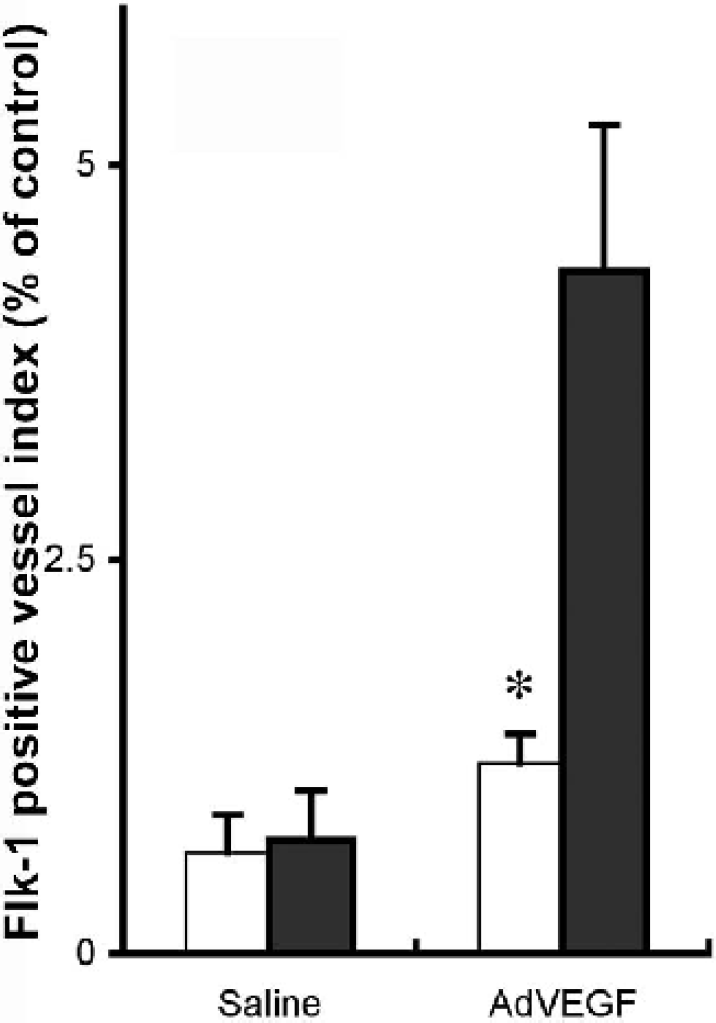
Bar graph shows the Flk-1 positive cell index examination in the Eng+/− and Eng+/+ mice after 3 weeks of saline or AdhVEGF transduction. Values are mean ± SD, N=9 per group. *P < 0.05, Flk-1 positive index is much lower in the Eng+/− mice compared with Eng+/+ mice.
TβR-1 expression
To identify the role of endoglin during angiogenesis, we examined brain TβR1 expression in the Eng+/− mice. Western blot analysis demonstrated that TβR-1 expression was comparable in the Eng+/− and Eng+/+ mice after 3 weeks of saline injection. Expression of TβR-1 increased after AdhVEGF transduction in both the Eng+/− and Eng+/+ mice. However, the increase of TβR-1 in the Eng+/− mice was greatly attenuated compared with the Eng+/+ mice (Fig. 5, P < 0.05). This result suggests that TβR-1 expression is closely related to endoglin levels.
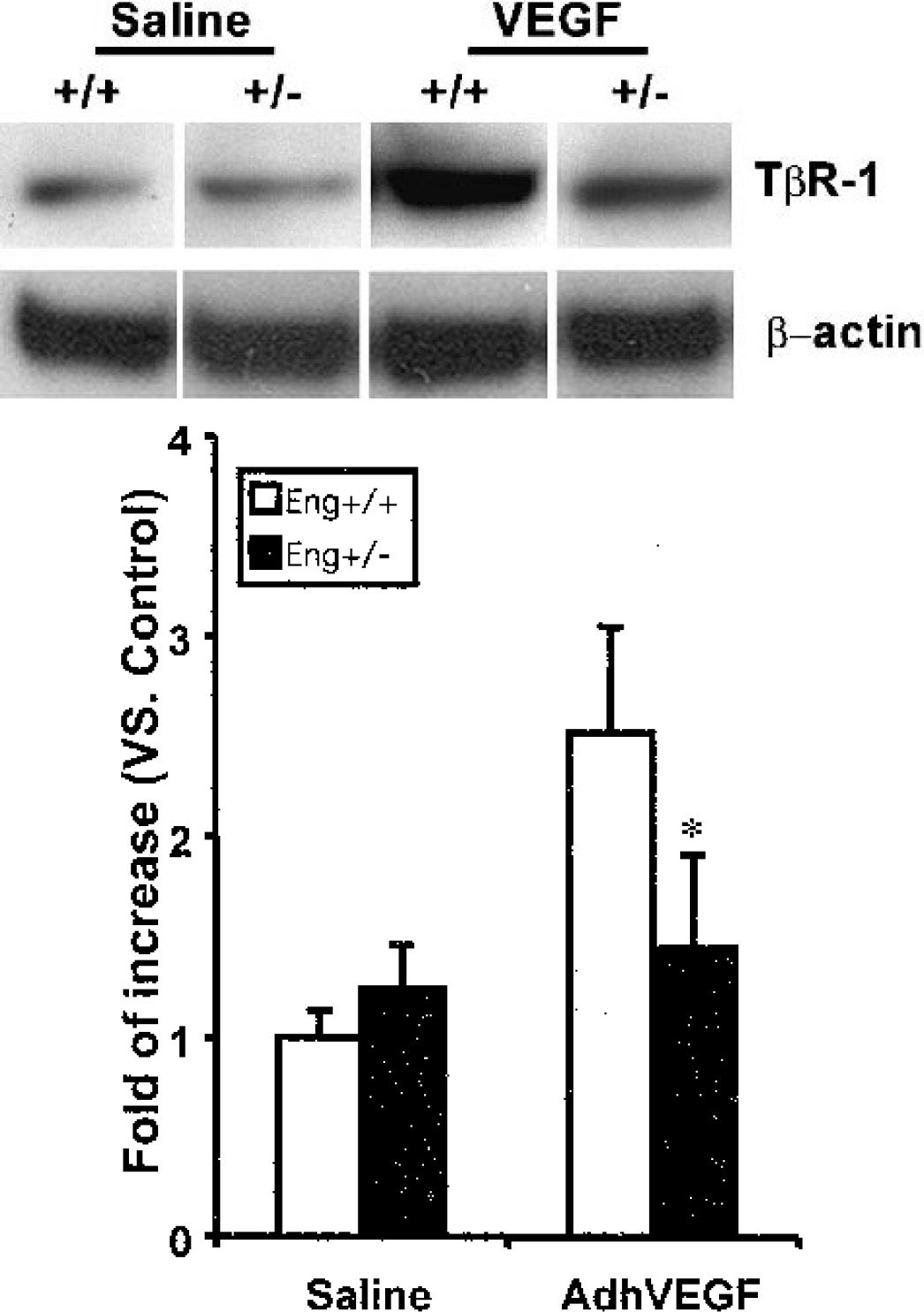
(Upper) One experiment of TGF-βR1 expression in Western blot analysis. TGF-βR1 presents in the major band at 55 kDa. TGF-βR1 is greatly increased both in the Eng+/+ and Eng+/− mice after 3 weeks of AdhVEGF transduction. (Lower) Bar graph shows that VEGF induced TGF-βR1 expression in the Eng+/+ and Eng+/− mice after 3 weeks of saline or AdhVEGF transduction. Values are mean ± SD, N = 3 per group. *P < 0.05, TGF-βR1 expression is attenuated in the Eng+/− mice compared with the Eng+/+ mice after AdhVEGF injection.-
Abnormal microvasculature in Eng+/− mice
DISCUSSION
The most important finding in this study is that VEGF hyperstimulation promotes angiogenesis to a similar degree in the adult endoglin haploinsufficient and wild type mice but that the incidence of abnormal microvessel formation in endoglin haploinsufficient mice is dramatically higher. Furthermore, we demonstrated that Flk-1 and TβR-1 expression was reduced in the Eng+/− mice, suggesting that haploinsufficiency of endoglin may down-regulate Flk-1 and TβR-1 activation. These changes may result in the development of abnormal microvessels in the Eng+/− mice.
We demonstrated that AdhVEGF transduction not only induces focal angiogenesis in the adult mouse brain but also promotes abnormal microvessel formation in the Eng+/− mice. This result provides a possible mechanism for the genesis of focal vascular malformations on the microscopic level. It may be consistent with the hypothesis that both mucocutaneous fragility and eventual formation of solid organ AVM in adult patients with HHT may be linked to a local increase in VEGF and associated signaling pathways (Guttmacher et al., 1995; Plauchu et al., 1989; Shovlin and Letarte, 1999). The variable HHT phenotype in the mouse models may reflect the wide range of disease severity in patients with HHT1, and, in almost all cases, only very localized sites are affected. It has therefore been proposed that other factors can locally precipitate the disease. This study shows that local VEGF release may be such a factor.
Mice carrying two independently derived targeted mutations in the endoglin gene have been extensively studied as a model of HHT1 (Bourdeau et al., 2001; Torsney et al., 2003). In both cases, the frequency of development of HHT-like bleeding vessels in Eng+/− mice was found to depend upon the background strain. The lesions developed at a much lower frequency and at a later age in the C57BL/6 background compared with 129/Ola. Furthermore, abnormal cerebral vessels have only been reported in C57BL/6mice aged 25 to 40 weeks (Satomi et al., 2003). We therefore used young (8 to 10 weeks old) Eng+/− mice in C57BL/6 background for our study because HHT had not been seen in young mice in this background. We also found that Eng+/− mice in the C57BL/6 background show no visible abnormal vascular structure or hemorrhage up to 12 weeks of age.
We demonstrated that a high incidence of abnormal microvessels occurred in eight of nine Eng+/− mice after AdhVEGF gene transduction but did not occur in the nine Eng+/+ mice, suggesting that the blood vessels in Eng+/− mice are abnormal in their response to VEGF, which indicates that during physiologic events that cause VEGF overexpression such as ischemia, inflammation, or wound repair, abnormal microvessels could develop.
Human HHT lesions are extremely variable and include dilated vessels, arteriovenous malformations, and capillary telangiectasias. However, it is not known how these abnormalities develop and what the starting point is. Cerebral lesions in HHT patients are detected only after they are symptomatic, whereas in the mouse model, we are able to investigate the development of the small abnormal vessels seen in this study. We have shown that AdhVEGF-induced angiogenic morphological changes occurred in the ipsilateral hemisphere in the Eng+/+ mice. These changes included angiogenic “sprouts,” tightly packed “glomerulold” structures, bridging, and dilated “mother vessels.” We also identified similar brain angiogenic changes in other species such as CD-1 mice after AdhVEGF transduction (Xu et al., 2003). However, VEGF overexpression induced different angiogenic changes in the Eng+/− mouse brain. These newly formed microvessels showed abnormal morphology such as a mass or single enlargement, clustered, twisted, or spiral microvessels. Confocal microscopy and three-dimensional reconstruction techniques enable clear identification of the abnormal microvasculature without having to resort to scanning electron microscopy (Satomi et al., 2003). These changes are similar to small cerebral AVMs in the adult HHT patient brain and may predispose the mice to brain hemorrhage.
There are two main features in patients with HHT: abnormal vessels and bleeding. Not all abnormal vessels bleed, but they can cause severe effects with relation to the affected organ. For example, dilated vessels in the brain may cause blood brain barrier leakage and edema. On the other hand, bleeding generally occurs from the abnormal vessels. Thus these two features of HHT are related but not the same. We have no evidence that these vessels in the mice are those that will bleed. We can only surmise that these small areas of abnormal morphology are likely to represent the beginning of a more mature nidus seen in patients.
The mechanism of abnormal microvessel formation in Eng+/− mouse brain is unclear. Flk-1 levels are reduced in Eng+/− mice, which is an important finding. During stable homeostasis, Flk-1 expression usually falls within with the normal range in the Eng+/+ mice (Shalaby et al., 1995). However, during VEGF hyperstimulation, endothelial cells and smooth muscle cells may be unable to produce sufficient Flk-1 for a normal response to the angiogenesis in the Eng+/− mice. As a result, cell proliferation and migration would be affected. In support of this hypothesis, the Eng+/− mice exhibit abnormal vessels with irregular layers of collagen and elastin, adding to the fragility of these vessels (Torsney et al., 2003). Unbalanced proliferation and migration of different cell type could cause newly formed microvessel instability and subsequent abnormal microvessel formation during angiogenesis.
Endoglin has been shown to be a negative modulator of TGF-β signaling (Lastres et al., 1996). Local endoglin levels may play a crucial role in the control of the TGF-β1 autoregulatory pathway. We found that TβR-1 expression was significantly reduced in the Eng+/− mice, suggesting that endoglin can control the TGF-β1 autoregulatory pathway. TGF-β1 is a critical cytokine that helps to stabilize and mature the newly formed microvessels in the adult mice. A decrease in either TGF-β1 or TGF-β1 receptors can lead to unstable cellular interactions in the vessel wall, dilated vessels, and vascular abnormalities. Such alterations could damage other angiogenic regulatory mechanisms and lead to deterioration of the vascular network associated with the progression of HHT (Bourdeau et al., 2000).
Despite these observations, it is still unclear how endoglin affects TGF-β signaling during angiogenesis and how this signaling pathway regulates newly formed microvessel maturation. Our results are consistent with the hypothesis that VEGF may play a role in the development of vascular malformations, although the exact mechanism by which this occurs remains to be determined. It is likely that factors other than the simple addition of VEGF contribute to the development of abnormal vessels because otherwise one might expect to see lesions in tissues where VEGF was normally upregulated during normal physiologic states, such as the endometrium during menses, or routine scar formation. We suggest instead that there are local tissue considerations that also influence the development of AVM.
CONCLUSION
We have demonstrated that VEGF hyperstimulation induces abnormal microvessels in the Eng+/− mouse brain but not in the brains of wild type animals. The Eng+/− mice appeared otherwise normal during the current observation period in the early stage of life. When challenged by environmental changes that upregulate angiogenic processes, endoglin haploinsufficiency appears to cause downregulation of the TGF-β1 pathway, resulting in abnormal microvessel formation. These results suggest that VEGF plays a role in the development of arteriovenous malformation in adult patients with HHT.