
Abstract
Secondary hypoxic/ischemic injuries, stemming from reductions in cerebral blood flow are important contributing factors in progressive neuronal dysfunction after brain trauma. A greater preclinical understanding of how brain trauma leads to secondary hypoxia/ischemia is necessary in the development of posttraumatic brain injury (TBI) therapeutics. To this end, we examined the density of microvascular coverage in the injured and contralateral cortical hemispheres using two intensities of fluid percussion trauma in rats. A silicone microangiography technique showed a significant loss in microvascular density in 2 atmosphere (atm) (16.9±3.8%) and 3 atm (15.7±1.3%) injured animals relative to sham animals (29.9±2.5%; P<0.01). RECA-1 immunohistochemistry indicated that capillary changes involved a reduction in capillary number and diameter. Reduction in microvascular density was shown to be a diffuse phenomenon occurring up to 4 mm rostral and caudal to the injury epicenter. Recovery of microvasculature occurred by 2 weeks after injury only in the 2 atm injury group. Expression of HIF1α and increased vascular endothelial growth factor expression were observed in the ipsilateral hippocampus suggesting sufficiently impaired microcirculation resulting in the expression of hypoxic-response proteins. Collectively, the results indicate diffuse and heterogeneous microvascular alterations as well as endogenous expression of neuroprotective and neovascularization pathways after TBI.
Introduction
Normal brain function is highly dependent on adequate cerebral perfusion and oxygen delivery. There exist well-defined thresholds of cerebral blood flow (CBF), which when compromised lead to impaired electrophysiologic activity, loss of consciousness, and at the extreme end, cell death and irreparable tissue loss (Verweij et al, 2007). In the context of traumatic brain injury (TBI), the importance of adequate blood and oxygen delivery becomes increasingly critical as reductions in CBF are associated with ischemia, low tissue oxygen tension (PaO2) and poor neurologic outcome (Bouma and Muizelaar, 1992; The Brain Trauma Foundation. The American Association of Neurological Surgeons. The Joint Section on Neurotrauma and Critical Care. Resuscitation of blood pressure and oxygenation, 2000). This suggests critical importance to the preservation of cerebral microvascular integrity and the corollary effect on adequate cerebral perfusion after TBI. However, this also implicates microvascular dysfunction or compromise as a key contributor to secondary complications including ischemia and increased intracranial pressure (Golding et al, 1999). Despite the strong correlation between microvascular dysfunction and TBI outcome, the mechanisms underlying vasospasm, stenosis, and microvascular loss resulting in hypoxia and ischemia are not well understood.
There exists ample clinical evidence indicating a reduction in global CBF after severe head trauma as well as data from animal models supporting this phenomenon (Shen et al, 2007; Yuan et al, 1988). A more detailed analysis of the regional distribution of microvascular compromise in response to varying levels of trauma would shed light on potential mechanisms to secondary brain ischemia and hypoxia. Furthermore, clarification of the focal and diffuse microvascular responses to injury could be correlated with the numerous and existing preclinical TBI studies describing neuronal populations susceptible to ischemic insult. From a biomechanical perspective tracing the extent of microvascular injury may elucidate information regarding the physical loading limits of microvasculature in response to tissue shearing and strain. Finally, description of the molecular signaling cascade associated with microvascular recovery in response to TBI may shed light on potential targets for treatment.
Several recent reports have showed an involvement of factors downstream of the prolyl hydroxylase domain 2 (PHD2)–hypoxia-inducible factor-1α (HIF1α) pathway in TBI. Regulation of HIF1α is mediated by a family of prolyl hydroxylases, with the PHD2 isoform being the predominant regulator of HIF1α stability under normoxia and mild hypoxia (Berra et al, 2003). Upregulation of vascular endothelial growth factor (VEGF) mRNA, protein and receptor expression have been reported (Dore-Duffy et al, 2007; Skold et al, 2005). VEGF expression has been shown to correlate with increased endothelial proliferation after injury (Morgan et al, 2007) whereas inhibition of its main receptor, VEGFR2, exacerbates neuronal and glial cell death (Skold et al, 2006). These data suggest not only a function for VEGF in neovascularization, but also in neuroprotection as has been documented in models of ischemia (Shen et al, 2006; Wang et al, 2007) and other systems (for reviews see Gora-Kupilas and Josko, 2005; Lazarovici et al, 2006).
There has been a greater appreciation for the diffuse pathophysiologic changes beyond the penumbra of the injury site associated with closed-head injuries. Diffuse secondary injury mechanisms including neuronal loss, axonal dysfunction, and impaired metabolic function have been described as occurring remote from the location of focal trauma (Farkas and Povlishock, 2007; Huh et al, 2008; Park et al, 2006a, 2006b; Wu et al, 2004). This study extends the recognition of diffuse injury mechanisms to include alterations to cerebral microvasculature. We examined the density of microvascular networks after two intensities of fluid percussion trauma using immunohistochemical and microangiographic techniques to identify capillary and microvessel changes, respectively. We report reductions in microvascular density in the ipsilateral cortex and hippocampus that were not limited to the region of focally applied trauma. The recovery of microvascular density was also dependent on the intensity of fluid percussion impact applied. We also show PHD protein degradation, the molecular consequence of which was stabilization of HIF1α and its downstream effector VEGF. Taken together these results identify a significantly hypoxic environment within the cortex and hippocampus after experimental TBI.
Materials and methods
All surgical procedures were performed in accordance with guidelines established by the Animal Care Committee at St. Michael's Hospital in accordance with the standards set by the Canadian Council on Animal Care.
Fluid Percussion Injury
We used the unilateral rat fluid percussion injury (FPI) model (Dixon et al, 1988; Dixon et al, 1987; McIntosh et al, 1989) in this study. In brief, male Sprague—Dawley rats (350 to 400 g) were anesthetized with 2.0% to 2.5% isoflurane delivered in compressed air. Temperature was maintained at 37°C by a thermal heating blanket. A craniotomy (∼2 mm diameter) was performed in the right lateral hemisphere, such that the medial edge of the craniotomy was approximately 2 mm from the midline suture, midway between bregma and lambda. A polyethylene tube with inner diameter ∼1.5 mm was fixed to the opening with cyanoacrylate adhesive and dental acrylic, filled with 0.9% isotonic saline and attached to the FPI device. Rats were subject to a moderate extradural FPI with the weighted pendulum set to an angle of 21.5° and 32° for 2 and 3 atm injuries, respectively. The duration of the waveform response due to fluid percussion was recorded as 12 to 15 ms. Bone wax was used to seal the hole in the skull and scalps sutured before recovery in a temperature-controlled chamber.
Immunohistochemistry
Coronal sections from 2 atm injured animals (n=4) were used for immunohistochemistry analysis of endothelial cell expression. Animals were anesthetized with ketamine and transcardially perfused with 0.9% saline followed by 4% paraformaldehyde. Extracted brains were postfixed overnight in 4% paraformaldehyde with 0.5 mol/L acetate solution before paraffin embedding. Serial coronal sections of 10 μm thickness were mounted on slides for immunofluorescent histochemistry. After deparaffinization sections were incubated with proteinase K for 15 mins and washed in 0.1 mol/L phosphate-buffered saline. Sections were blocked for 1 h in 0.1 mol/L phosphate-buffered saline containing 10% normal goat serum and 0.1% Triton-X. Primary incubation of mouse anti-RECA-1 antibodies (Abcam, Cambridge, MA, USA), a marker for endothelial cells, was performed at a dilution of 1:500 in blocking solution overnight at 4°C. Secondary incubation was performed with Alexa 488 goat anti-mouse antibodies (Molecular Probes, Burlington, ON, Canada) for 1 h at room temperature in blocking solution at a dilution of 1:1,000. Three washes were made in 0.1 mol/L phosphate-buffered saline between each step. Negative controls were run simultaneously with the omission of primary antisera.
Silicone Perfusion Microangiography
Treatment groups used for microangiography studies included sham (n=6), 2 atm FPI (n=10), and 3 atm FPI (n=10). Survival time points for 2 and 3 atm injury groups were 24 h and 14 days after fluid percussion trauma. Animals were anesthetized with ketamine and transcardially perfused with isotonic saline for 2 to 3 mins. A blue silicone compound (MV-120; Flow Tech Inc., Carver, MA, USA) was injected at an initial rate of 180 mL/h for 5 mL then at 55 mL/h for a volume of 10 mL with a KDS120 syringe pump (KD Scientific, Holliston, MA, USA). The silicone compound was cured for a minimum of 90 mins before brain extraction and storage in 70% ethanol. After graded alcohol dehydration, brains were transferred to methyl salicylate for tissue clearing. A brain slicer (Zivic Instruments, Pittsburg, PA, USA) producing 2 mm coronal slices was used to section the cerebral cortex. Slices were mounted on slides, coverslipped, and imaged with a Nikon Eclipse 90i microscope.
Western Blotting
Brain tissue samples were collected from the injury epicenter and surrounding tissue of the cerebral cortex (approximately 2 mm3), the contralateral cortex as well as the ipsilateral and contralateral hippocampus from sham animals and 24 h postinjury animals (n=4 per treatment group; n=5 for VEGF blots). Samples were homogenized in lysis buffer containing protease inhibitors (50 mmol/L Tris-HCl, 1% NP-40, 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L phenylmethylsulphonyl fluoride, 1 μg/mL aprotinin, 1 μg/mL leupeptin, 1 μg/mL pepstatin). Protein quantification was determined by the modified Lowry method (Peterson, 1977). Samples were normalized for equal loading (20 μg per lane) and were electrophoresed on 7.5% or 12% SDS—polyacrylamide gels and transferred overnight to nitrocellulose membranes. Blocking of membranes was performed in 5% nonfat milk blocking solution for 1 h at room temperature. Immunoblots were probed for PHD2 and HIF1α with rabbit antipolyclonal PHD2 (Novus Biologicals, Littleton, CO, USA) and monoclonal anti-HIF1α antibodies (Millipore, Billerica, MA, USA), respectively. VEGF-A expression was assessed with rabbit polyclonal anti-VEGF-A antibodies (Santa Cruz, Santa Cruz, CA, USA). Anti-PHD2 incubations were performed at 1:800 dilution in blocking solution. Dilution of anti-HIF1α was 1:1,000 in blocking solution. Anti-VEGF-A incubations were performed at 1:200 in blocking solution. All primary incubations were performed overnight at 4°C. Secondary antibody incubation was performed for 1 h with horseradish peroxidase-conjugated goat anti-rabbit or goat anti-mouse (1:3,000 dilution) secondary antibodies. Rabbit polyclonal anti-ERK1,2 antibodies (Sigma, Oakville, ON, USA) were used as a loading control (1:15,000 dilution) and visualized with horseradish peroxidase-conjugated goat-anti-rabbit (1:3,000 dilution) secondary antibodies. Washes in three changes of Tris-buffered saline Tween-20 were performed between incubations. Bands were visualized using a chemiluminescence kit and exposure to X-ray film.
Image Analysis
Immunohistochemistry analysis of RECA-1 labeling included quantification of the diameter and the number of RECA-1-positive objects captured within a sampling field in the cerebral cortex. Sections were sampled from regions approximately 3 mm rostral and caudal to the injury site as well as at the injury epicenter. Four sampling fields per region were captured on a × 20 objective using a Nikon Eclipse 90i microscope. For sections taken from the injury epicenter, sampling fields were chosen off-axis to the injury epicenter due to excessive background labeling that could contribute to false positives. Capillaries were identified as RECA-1-positive objects with diameters of less than 10 μm.
For microangiography studies, slice images were captured with an extended depth of focus software module (Nikon, Toronto, ON, Canada) which allowed in-focus image planes taken at 150 μm intervals to be projected as a single-focused image representative of a 2-mm thick brain section. Image J (http://rsb.info.nih.gov/ij/) was used for microvascular density analysis. Sampling fields with dimensions 2.3 mm (width) × 1 mm (height) were taken from the ipsilateral and contralateral cortex and hippocampus regions. Each brain yielded approximately 5 brain sections encompassing an area 4 mm rostral and 4 mm caudal to the injury epicenter. Analysis of sections on a × 10 objective indicated that the silicone dye permeated microvasculature with diameters as small as 15 μm.
Statistical Analysis
Sampling field values for each of the regions were averaged to give a representative value for that region. The diameter and number of RECA-1-positive objects were compared between the injured ipsilateral and contralateral cortex using a t-test. A subanalysis was performed using a t-test to examine the diameter and number of RECA-1-positive objects at 3 mm rostral and caudal to the injury epicenter, as well as at the injury epicenter.
A two-way repeated measures analysis of variance was used to compare the mean density of cortical microvasculature in sham, 2 and 3 atm injured animals in both the injured and contralateral hemispheres at 24 h and 2 weeks after injury. Post hoc analysis of pair-wise comparisons was performed using the Holm—Sidak method. A subanalysis of microvascular density from serial sections within injury groups was performed using a one-way analysis of variance to examine the relative changes in microvascular density occurring rostral—caudal to the injury epicenter. Error bars represent mean standard errors. Significance was assumed at a P-value≤0.05.
Results
Reduced Capillary Density and Reduction in Diameter
The mean number of RECA-1 objects counted in the hemisphere contralateral to the injury site was 44. 3±3.8 and 28.3±2.8 in the injured cortex (P=0.002; Figures 1A–1C inset). A further subanalysis of RECA-1 expression indicated a significant reduction in RECA-1 objects 3 mm caudal to the injury site with 55.9±3.2 objects in the contralateral cortex and 25.3±2.8 objects in the injured cortex (P<0.001; Figure 1C). Off-axis sample fields from the injury epicenter indicated a reduction in RECA-1 expression, but were not statistically significant. Values were 41.8±6.5 objects in the contralateral cortex and 29.6±5.8 objects in the injured cortex (P=0.21).
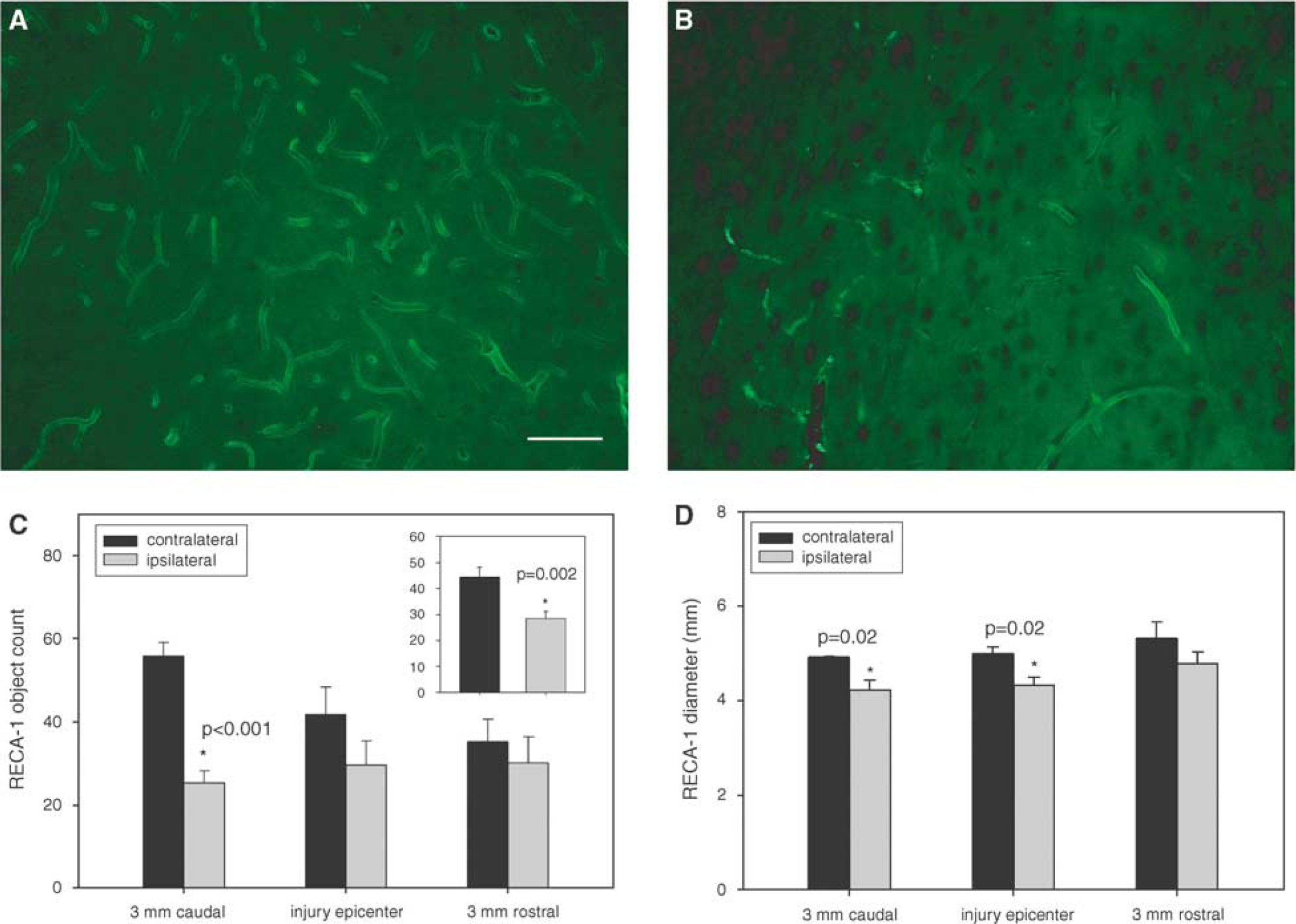
Representative image of RECA-1 labeling in the (
The mean diameters of RECA-1-positive capillaries in the hemisphere contralateral to the injury were 5.08±0.13 μm. Mean diameters in the injured cortex were 4.44±0.14 μm (P=0.002; Figure 1D). Analysis of capillary diameter by sampling region indicated a nonsignificant difference 3 mm rostral to the injury epicenter with mean capillary diameters of 5.32±0.34 and 4.78±0.25 μm in the contralateral and ipsilateral cortices, respectively (P=0.25). At the injury epicenter, mean diameters were significantly smaller in the injured cortex with mean diameters of 4.33±0.17 versus 4.99±0.15 μm in the contralateral cortex (P=0.02). Values 3 mm posterior to the injury epicenter were also significantly different with diameters 4.21±0.22 μm in the injured cortex and 4.92±0.02 μm in the contralateral cortex (P=0.02).
Reduced Perfusion of Cerebral Micovessels
The density of microvascular networks in the cortex was significantly different (P=0.01) at 24 h and 2 weeks after injury between sham, 2 and 3 atm injury groups. There were no significant differences in microvascular density between the cortices contralateral to the injury in 2 and 3 atm injured animals or the left hemisphere of sham animals. At 24 h after injury the mean microvascular density in the right cerebral cortex of sham animals was 29.9±2.5% area coverage, 16.9±3.8% in the 2 atm injury group and 15.7±1.3% in the 3 atm injury group (Figures 2A and 2B). The density values of the injured cortex decreased significantly in 2 and 3 atm injury groups relative to sham animals (P=0.003 and 0.001, respectively; Figures 2C and 2D). There was no significant difference in density values between 2 and 3 atm injured cortices at 24 h after injury.
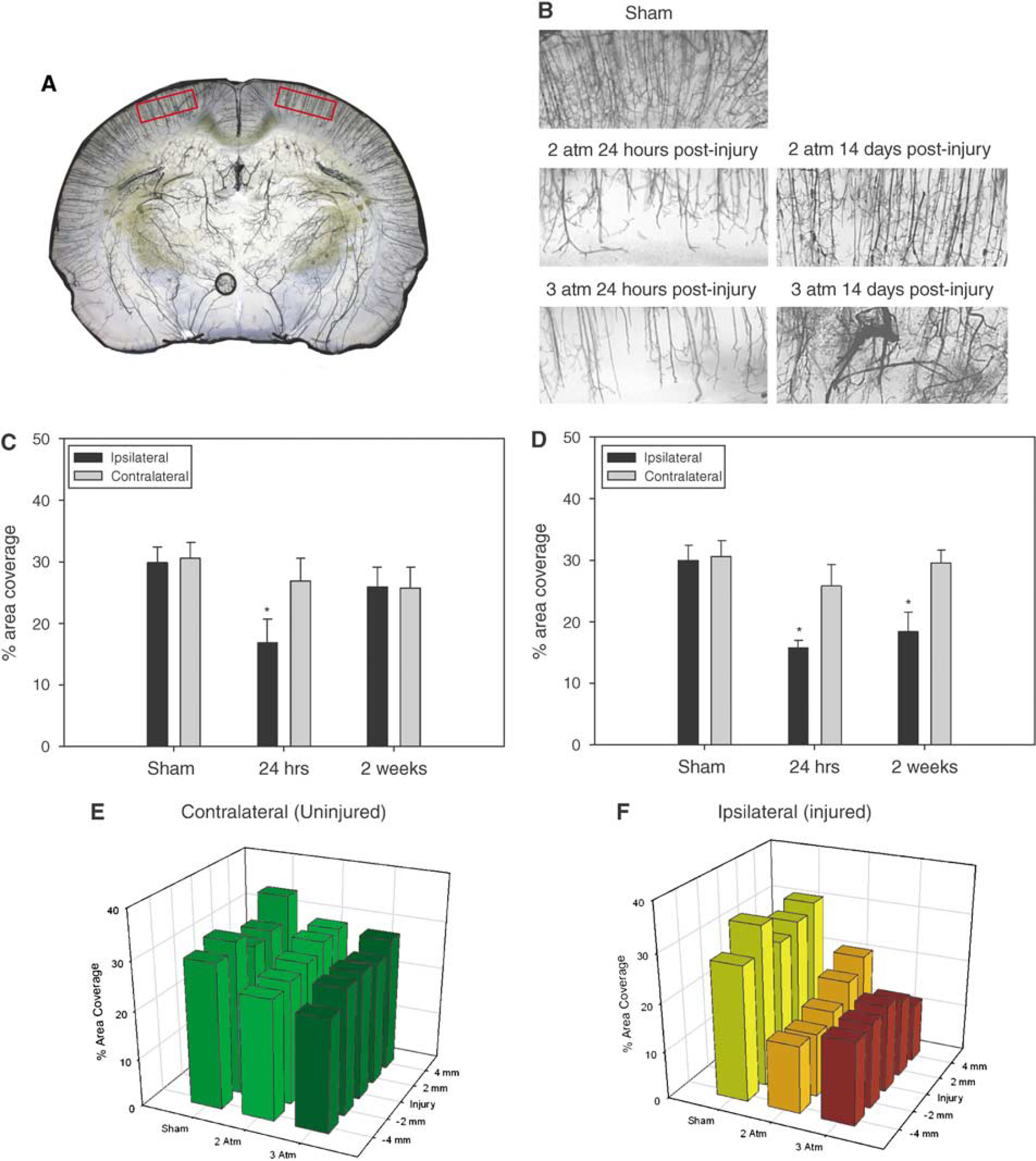
(
At 2 weeks after injury, the density of perfused microvasculature in the 2 atm injured cortex suggested recovery to near-sham levels (Figure 2C). The mean value of microvascular coverage at 2 weeks after injury in the 2 atm group was 25.9±3.2%. There was no significant difference between the right cerebral cortex of sham animals relative to the cortices of 2 atm injury animals. There was also no significant difference between the injured and contralateral cortices of 2 atm injured animals at 2 weeks after injury. By contrast, microvascular density in the injured cortex of the 3 atm injury group remained significantly less (P=0.011) at 18.4±3.1% compared with sham density values (Figure 2D). There was also a significant difference in the microvascular density between the injured and contralateral cortices of 3 atm injured animals at 2 weeks after injury (P=0.001).
There was a significant difference in microvascular density among treatment groups and hemispheres in the hippocampus (P=0.004). Post hoc analysis indicated no significant difference in perfused microvascular density in the 2 atm injury group at any time point (Figures 3A–3C). Interestingly, in the 3 atm injury group there was notably less microvascular coverage in the contralateral hemisphere relative to the injured hemisphere at 24 h and 2 weeks after injury. A post hoc comparison of ipsilateral and contralateral hippocampus regions indicated a significant difference in microvascular density at 24 h (P=0.002) and at 2 weeks after injury (P=0.001; Figures 3B and 3D). Despite the difference in bilateral perfusion of the hippocampus the observed density levels were not significantly different from sham density levels.
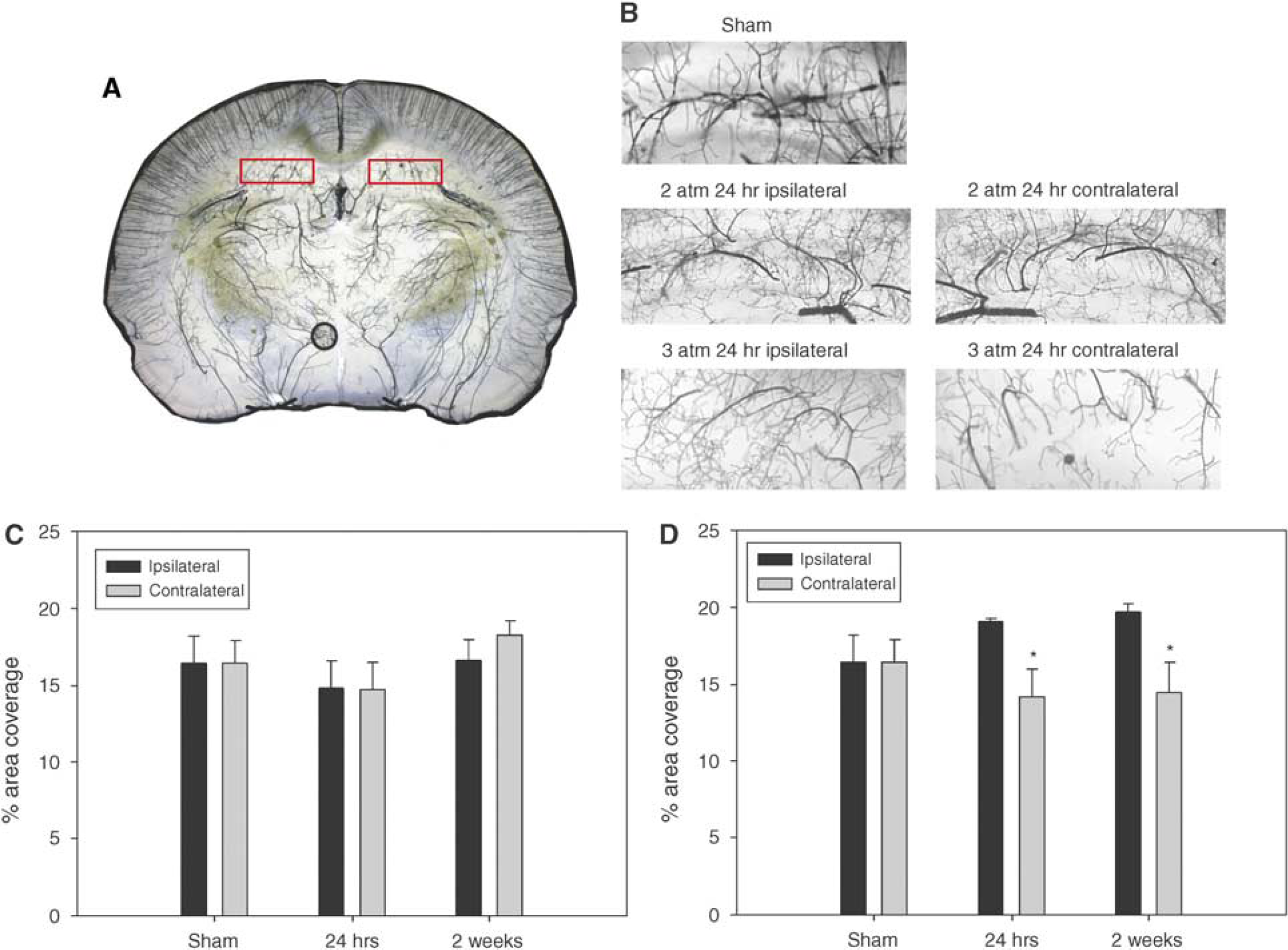
(
Expression of Hypoxic Signaling Proteins
The lower capillary count, decreased diameter and the reduction in perfused microvasculature suggested the possibility of a hypoxic tissue response. Accordingly, we examined the induction of a hypoxia-mediated pathway involving PHD2, HIF1α, and VEGF-A. Expression of these proteins was evaluated in sham animals and at 24 h after injury in 2 atm injured animals. Constitutive expression of PHD2 was observed in the cortex and hippocampus of sham animals (Figure 4A). Interestingly, PHD2 was detected in neither the injured nor the contralateral cerebral cortex or hippocampus, however, breakdown fragments were observed at lower molecular weights (∼40 kDa). Expression of HIF1α was detected in the injured hippocampus but only ipsilateral to the injured hemisphere (Figure 4B). We were unable to detect HIF1α expression in the cortex (data not shown). Finally, we examined expression of VEGF-A—which displays HIF1α-dependent transcription under hypoxic conditions—in sham and injured animals. We observed an increase in VEGF-A expression in the hippocampus (Figure 4C) whereas expression in the injured cortex was similar to levels in sham animals (data not shown).
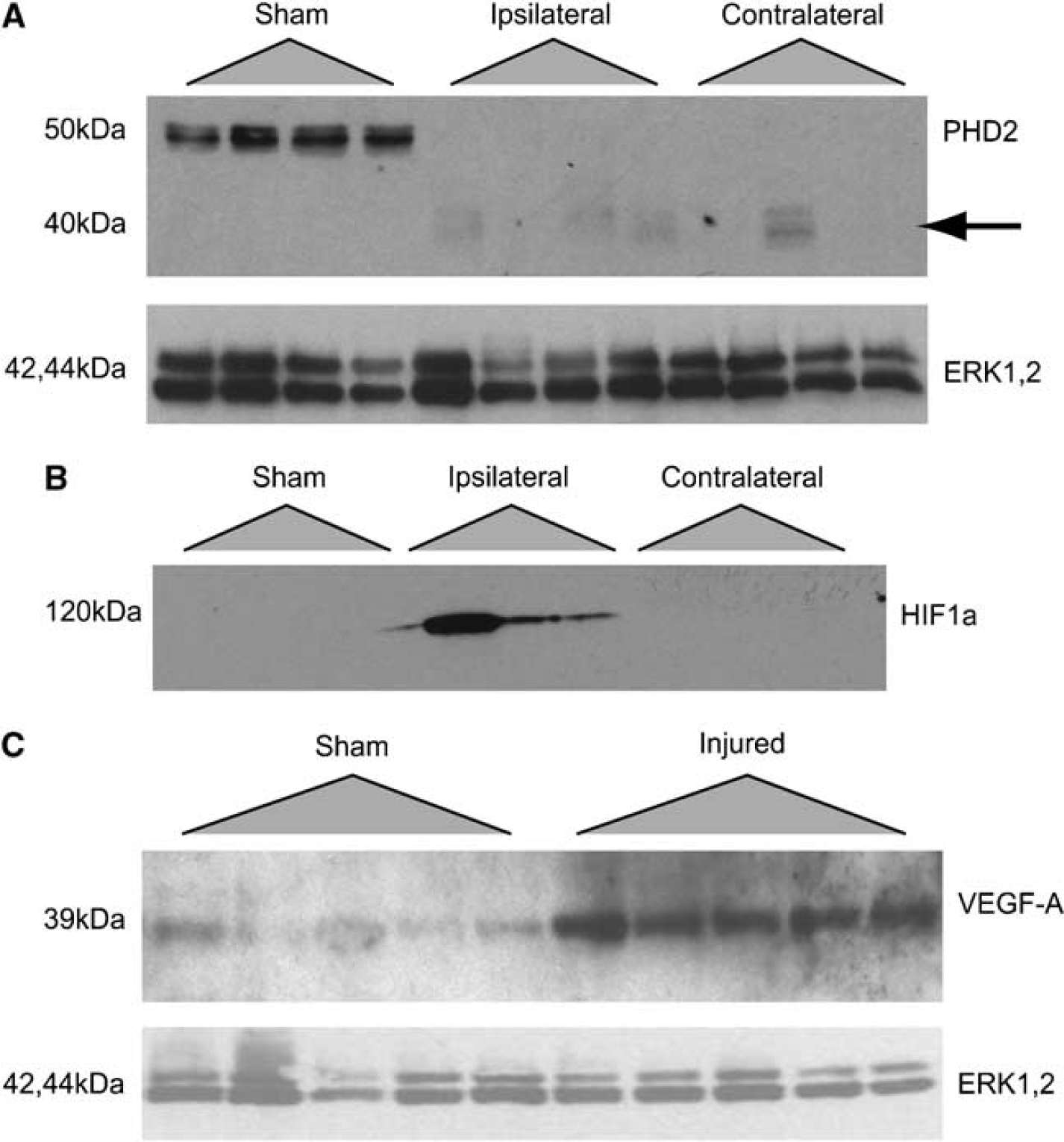
(
Discussion
The consequences of microcirculatory impairment manifest as decreased CBF and reduced tissue oxygen delivery after TBI. These phenomena are well-documented clinical findings with significant implications for outcome after brain trauma (for review see Golding et al, 1999) evolving into complication risks including ischemia, hypotension (Longhi et al, 2007), and increased intracranial pressure (Vespa, 2003). However, the regional distribution of these microvascular changes as contributors to global reductions in CBF are not readily apparent. Moreover, the extent to which microvascular changes are due to an imbalanced physiologic response mechanisms (e.g. vasoconstriction) or due to mechanical perturbation of the microcirculation requires further clarification.
In this study we sought to determine a relationship between biomechanical loading on the brain and microvascular response using a TBI model of fluid percussion trauma. The observed decreases in capillary diameter and density suggested the potential for an ischemic or hypoxic response to fluid percussion trauma. We examined expression of proteins known to be involved in a hypoxia-mediated signaling pathway and observed increased expression of VEGF-A suggesting the possibility of an endogenous microvascular reparative response mechanism.
The quantitative RECA-1 immunohistochemistry data suggested both a loss of capillary networks as well as decrease in overall capillary diameter. The reduction in RECA-1 labeling after TBI is consistent with a previous report in which microvascular basal lamina loss was indicative of microvascular loss after TBI (Muellner et al, 2003). This is further corroborated by a study showing a loss of endothelin markers indicating loss of microvasculature after TBI (Lin et al, 2001). The reduction in capillary density as well as the decreased density of microvascular networks as assessed by silicone perfusion also indicates that vasoconstriction is involved in this model of TBI as well. Several recent studies have examined potential mechanisms involved in the vasoconstrictive response to trauma. Proposed mediators of the microvascular contractility in TBI include calponin (Kreipke et al, 2006; Kreipke et al, 2007),
The recovery of the silicone perfusion density at 2 weeks after injury has several implications about mechanisms of microvascular disturbance after TBI. Recovery of microvascular density in the 2 atm injury group suggests that vasoconstrictive events resolved by 2 weeks after injury. Furthermore, given the significant loss in RECA-1 capillary expression at 24 h after injury and the subsequent recovery of density at 2 weeks after injury, there exists the possibility of the involvement of neovascularization processes as well.
Mammalian cellular oxygen sensing is accomplished by the iron- and 2-oxoglutarate-dependent PHD dioxygenases. PHD proteins regulate the oxygen-dependent proline hydroxylation of HIF1α, which leads to HIF ubiquitination, association with the von Hippel-Lindau and its E3 ubiquitin ligase complex and subsequent proteosomal digestion (Berra et al, 2006; Jaakkola et al, 2001; Pugh et al, 1999; Ratcliffe et al, 1998). The expression of HIF under normoxic conditions is short lived, as there is abundant oxygen necessary for PHD-dependent HIF hydroxylation. HIF is stabilized, however, under hypoxic settings (Pugh et al, 1999) where it mediates the transcription of many genes involved in the cellular adaptation to hypoxia (e.g., VEGF, erythropoietin, and glucose transporter protein 1). Although there exists a family of prolyl hydroxylase-negative regulators of HIF (PHD1 to PHD3), it is generally accepted that PHD2 is the critical oxygen sensor regulating the stability of HIF1α protein (for review see Berra et al, 2006).
The loss of PHD2 expression and the increased expression of HIF1α and VEGF in the injured cerebral hemisphere supports the possibility of induction of neovascularization mechanisms after TBI. Interestingly, the absence of PHD2 expression was observed in both the contralateral and ipsilateral cortices, whereas HIF1α expression was only detected in the injured side of the brain. This suggests an alternate signaling mechanism in the contralateral cortex that does not result in the stabilization of HIF. Furthermore, HIF1α and VEGF expression in injured tissues was largely localized to the hippocampus of the injured side. This is in contrast to silicone perfusion data that indicated a reduction, though not statistically significant, at 24 h after injury. It is possible that the lack of HIF1α and VEGF expression in the cortex represents temporal effects of expression that were not detected in our sampling time point. Collectively, however, these data suggest that the microvascular response to trauma differs between the cortex and the hippocampus. Furthermore VEGF activity may be involved in region-specific functions such as neuroprotection in the hippocampus while playing a more predominant function in neovascularization in the cerebral cortex. It should be noted, however, that despite implication of VEGF as a mediator of neovascularization and neuroprotection, some evidence also suggests that VEGF expression may be a cause of edema after TBI (Chodobski et al, 2003). Clearly, the multifaceted function of VEGF requires further clarification in the context of TBI.
The microvascular response to injury also demonstrated heterogeneity with respect to the degree of trauma sustained. Although the 2 and 3 atm injury groups had similar levels of microvascular reduction at 24 h after injury, there was little discernable recovery of microvascular density in the 3 atm injury group at 2 weeks after injury. This suggests that the extent of injury at the higher level of tissue trauma perhaps resulted in greater microvascular loss than was salvageable through recovery of microvascular tone or neovascularization. This dose-dependent effect between microvascular recovery and injury severity indicates a clear pathobiologic response as a function of trauma severity. It is interesting to note that the recovery of microvasculature correlates with the partial spontaneous recovery of corpus callosum function reported in several TBI studies (Baker et al, 2002; Reeves et al, 2005), further strengthening the link between neurophysiologic function and cerebral microvasculature.
Finally, the distribution of RECA-1 labeling as well as the reduction in microvascular density remote from the injury epicenter extends the concept of diffuse injury mechanisms to include microvascular disturbances as well. Our results indicate that reductions in microvascular density extended both rostral and caudal to the injury site, with a preference for capillary loss caudal to the injury site. Furthermore, in the 3 atm injury group there was a curious finding of decreased microvascular density in the contralateral hippocampus relative to the side sustained the fluid percussive impact. This seemingly counterintuitive response may represent a compensatory physiologic mechanism in severe TBI whereby perfusion is increased to the injured side at the expense of the uninjured contralateral hippocampus. An alternative explanation may be related to biomechanical loading on the brain with severe brain trauma. Compression of the hippocampus on the injured side may result in shearing or expansion on surrounding tissue regions such as the contralateral hippocampus resulting in a loss or injury to the microvasculature.
Conclusion
The cerebral microvascular response to brain trauma continues to be recognized as an important initiator of secondary injury complications and contributor to poor neurologic outcome. Although reductions in global CBF after TBI are well documented, the regional distribution of underlying microvascular changes responsible for posttraumatic reduction in CBF is not well defined. In this study we showed the diffuse and heterogeneous distribution of the microvascular response and recovery after FPI in the rat. We also showed activation of a hypoxia-mediated signaling pathway resulting in the expression of VEGF. The precise function of VEGF in TBI and its function in microvascular recovery and neuroprotection remain to be clarified. It has become increasingly evident that the complexity of the pathophysiologic response to brain trauma extends beyond the penumbra of the injured tissue and also involves dysfunction of microvasculature. The relationship of microvascular dysfunction and its contribution to neuronal and white matter deterioration or vice versa warrants further investigation.