
Abstract
The distribution and time course of expression of the heat shock/stress proteins, hsp27 and hsp72, were evaluated in a highly controlled gerbil model of ischemic injury and tolerance induction, in which the duration of ischemic depolarization in each hippocampus provides a precise quantitative index of insult severity. Gerbils were subjected to brief priming insults (2- to 3.5-minute depolarization) that produce optimal preconditioning, to severe test insults (6- to 8.5-minute depolarization) that produce complete CA1 neuron loss in naive animals, or to combined insults administered 1 week apart, after which almost complete tolerance to CA1 neuron injury is observed. Immunoreactivities of hsp27, hsp72, glial fibrillary acidic protein and microtubule-associated protein 2 (MAP2) were evaluated in animals perfused at defined intervals after the final insult in each treatment group, using a variation of established antigen-retrieval procedures that significantly improves detection of many proteins in vibratome brain sections. Hsp72 was detected in CA1 neurons of some hippocampi 2 to 4 days after preconditioning, but this was only seen after the longest priming depolarizations, whereas shorter insults that still induced optimal tolerance failed to induce hsp72. Hsp72 was induced after test insults in preconditioned hippocampi, but at a higher depolarization threshold than observed for naive animals. An astrocytic localization of hsp27 was observed in regions of neuron injury, as indicated by reduced MAP2 immunoreactivity, and was primarily restricted to dentate hilus after preconditioning insults. These results establish that limited hilar lesions are characteristic of optimal preconditioning, whereas prior neuronal expression of either hsp72 or hsp27 is not required for ischemic tolerance.
Dynamic changes in gene expression occur after transient brain ischemia, including the induction of several heat shock or stress proteins (Koistinaho and Hökfelt, 1997; Nowak and Kiessling, 1999). These are generally considered to play adaptive, protective roles in diverse injury models (Abe and Nowak, 1996b; Kirino 2002; Parsell and Lindquist, 1994; Sharp et al., 1999). Such responses have received particular attention in the context of ischemic tolerance, with many studies documenting neuronal induction of the 70-kd stress protein, hsp72, after brief insults that provide protection against subsequent challenges (Chen et al., 1996; Glazier et al., 1994; Kirino et al., 1991; Liu et al., 1993; Nishi et al., 1993; Simon et al., 1991). Although recent results indicate that prior hsp72 mRNA induction is not required for preconditioning (Abe and Nowak 1996a, 2000, 2004), some evidence suggests that tolerant neurons show facilitated hsp72 mRNA and protein expression after subsequent test insults (Aoki et al., 1993; Furuta et al., 1993; Kanemitsu et al., 1994).
Postischemic responses of nonneuronal cell types have also been reported in the context of preconditioning. Notably, the small heat shock protein, hsp27, exhibits increased expression after ischemia (Higashi et al., 1994; Kato et al., 1995; Kato et al., 1994b; Valentin et al., 2001), with a largely astrocytic localization after global or focal ischemia (Kato et al., 1995; Kato et al., 1994b). However, transient expression of hsp27 has also been reported in hippocampal CA1 neurons after priming insults, suggesting involvement in preconditioning (Kato et al., 1994b), and recent studies support a role for hsp27 expression in survival of dorsal root ganglion neurons in vivo and in vitro (Costigan et al., 1998; Lewis et al., 1999). Astrocyte activation, evaluated by morphologic changes in glial fibrillary acidic protein (GFAP), can occur in some hippocampal regions even after mild insults that induce tolerance (Kato et al., 1994a), prompting the suggestion that responses in this cell type could contribute to neuron protection. Conversely, prominent glial activation in dentate hilus after preconditioning insults may be secondary to injury of the exceptionally vulnerable somatostatin-containing neurons in this region (Matsuyama et al., 1993), although other results indicate that hilar lesions do not necessarily accompany preconditioning (Sugimoto et al., 1993).
The present studies were designed to resolve these controversies regarding the involvement of hsp27 and hsp72 protein expression in ischemic preconditioning. Recent studies from this and other laboratories have used a highly reproducible gerbil ischemia model in which microelectrodes record ischemic depolarization in each hippocampus, allowing the definition of quantitative depolarization thresholds for neuron injury and tolerance induction (Abe and Nowak, 1996a, 2004; Sorimachi et al., 2002). The reproducibility of this model permits changes in diverse parameters to be compared among animals, and at various recirculation intervals after priming insults, with a high degree of confidence in the histopathological outcome that can be expected in a given hippocampus. The present studies characterized the distribution and time course of hsp72 and hsp27 protein expression after preconditioning, as well as after test insults in naive and preconditioned animals. These stress-induced responses were compared with changes in microtubule-associated protein 2 (MAP2) and GFAP as indicators of neuron injury and glial activation, respectively.
MATERIALS AND METHODS
Ischemia model
All studies used a quantitative gerbil bilateral common carotid artery occlusion model that incorporated hippocampal DC potential recording to monitor the duration of ischemic depolarization during each insult (Abe and Nowak, 1996a, in press). Female Mongolian gerbils weighing 50 to 75 g were obtained from Charles River Lab Animals (West Brookefield, MA, U.S.A.). All procedures were performed according to a protocol approved by the Animal Care and Use Committee, University of Tennessee. Each gerbil was anesthetized with 2% to 2.5% halothane in a mixture of 30% O2 and 70% N2. The common carotid arteries were exposed and looped with suture, and the animal was then fixed in a stereotaxic frame in a prone position. The scalp was exposed and burr holes made for an epidural temperature probe (Physitemp Instruments, Clifton, NJ, U.S.A.) and for recording electrodes. A glass electrode filled with 2-mol/L sodium chloride (resistance 1 to 5 MΩ, 50 μm diameter) was placed in each hippocampus. DC potentials were recorded continuously using a battery powered amplifier (Electro 705, World Precision Instruments Inc, Sarasota, FL, U.S.A.) and a digital interface (SuperScope II, GW Instruments, Somerville, MA, U.S.A.). Ischemia of varied durations was produced by applying tension to the suture loop, thereby occluding the carotid arteries. Throughout these procedures, rectal and epidural temperatures were separately maintained at 37°C by means of independent heating lamps under feedback control. Electrodes were removed immediately after recirculation, and the scalp and neck incisions were closed, after which anesthesia and temperature control were maintained for an additional 90 minutes.
Durations of carotid artery occlusion were routinely chosen to produce two distinct ranges of insult severity, based on prior studies (Abe and Nowak, 2004) and confirmed in preliminary experiments described below. Priming insults resulted in hippocampal depolarizations of 2 to 3.5 minutes, shown to induce optimal preconditioning. Test insults produced 6- to 8.5-minute depolarizations, which reproducibly destroyed CA1 neurons in naive animals. A third group experienced priming insults followed by test insults 1 week later. Individual hippocampi that fell outside the indicated depolarization ranges were excluded from the grouped data. Sham animals were subjected to surgery and electrode placement without carotid occlusion.
Histopathology
Ischemic thresholds for injury and tolerance were defined in an initial group of animals (n = 47) subjected to single or repeated insults of varied duration. These animals were perfused after 7 days with 10% formalin, 10% acetic acid, and 80% methanol, after which brains were embedded in paraffin. Sections (8 μm) were cut on a rotary microtome, mounted on glass slides and stained with hematoxylin/eosin. Neuron counts in CA1 at the level of dorsal hippocampus were obtained at 250X magnification with a calibrated reticle and expressed as cells per millimeter of pyramidal layer arc, as previously described (Abe and Nowak, 2000).
Immunocytochemistry
Animals subjected to defined intervals of priming, test, or repeated ischemia (n = 98) were perfusion fixed with 4% paraformaldehyde in 100-mmol/L sodium phosphate (pH 7.4) at recirculation intervals of 6 hours and 1, 2, 4, and 7 days after the final insult. Sham-operated animals corresponding to both single and repeated ischemia groups were perfused 7 days after the final surgery. At each time point three to seven animals were perfused from each group, yielding six to 12 hippocampi that met the depolarization criteria for inclusion. Vibratome sections (50 μm) were cut and stored refrigerated in 10-mmol/L sodium phosphate-buffered saline (PBS) containing 0.02% sodium azide. Immunocytochemistry for hsp27, hsp72, MAP2, and GFAP were performed by modifications of published procedures (Vass et al., 1988). In the case of hsp72, and to some extent for MAP2 and GFAP, detection was significantly enhanced by the use of an antigen-retrieval procedure (detailed below), and this was routinely included as a pretreatment for detection of these proteins. Thereafter the immunocytochemistry procedure was identical for all antigens. Sections were incubated for 30 minutes in 50-mmol/L NH4Cl in PBS and for 1 hour in 0.3% Triton X-100 in PBS. After blocking for 3 hours in 4% bovine serum albumin in 100% normal goat serum, sections were incubated overnight at 4°C in primary antibodies prepared in a 10-fold dilution of the blocking solution. Antibodies and dilutions used were as follows: hsp27, rabbit polyclonal SPA-801 (StressGen Biotechnologies Corp, BC, Canada; 1/1000); hsp72, mouse monoclonal SPA-810 (StressGen; 1/400); MAP2, mouse monoclonal M-4403 (Sigma Chemical, St. Louis, MO, U.S.A.; 1/500); and GFAP, mouse monoclonal RPN-1106 (Amersham Pharmacia Biotech, Piscataway, NJ, U.S.A.; 1/200). After three washes in PBS, endogenous peroxidase activity was quenched in 3% H2O2, 10% methanol in PBS. Sections were washed several times in diluent and incubated for 2 hours at room temperature in peroxidase-labeled antimouse or antirabbit secondary antibodies (1/200 dilutions of product numbers 115-035-146 and 111-035-144, respectively, Jackson ImmunoResearch, West Grove, PA, U.S.A.) that had been preabsorbed overnight with five volumes of normal gerbil serum prior to dilution. After several washes in PBS, immunoreactivity was detected by incubation with 0.5- mg/mL 3,3'-diaminobenzidine, 0.015% H2O2 and 0.1% nickel sulfate in PBS. Sections were then rinsed in 50-mmol/L sodium phosphate, mounted on gelatin-coated slides, dehydrated through graded alcohol and xylene, and coverslipped. For some comparisons, immunocytochemical signal intensity was graded on the following scale: 0, no signal; 1, detectable above background; 2, moderate or scattered; 3, intense and homogeneous.
Antigen retrieval
Several antigen-retrieval methods have been used to unmask antigens in paraffin sections (Cattoretti et al., 1993; Shi et al., 1991), and recent results suggest that such approaches can also be useful in frozen and vibratome sections in which antigen detection has not been a generally recognized problem (Jiao et al., 1999; Shiurba et al., 1998). Heating in citrate buffer improved hsp72 detection in preliminary experiments (Nishino et al., 1998), but evidence that calcium chelation contributes to antigen retrieval (Morgan et al., 1997) suggested the more reliable approach used in the present studies. Floating sections were heated in a water bath at 80°C for 20 minutes in glass vials containing 2 mL of 1-mmol/L EGTA (pH 8.5), with inclusion of 0.3-mol/L sucrose to maintain osmolarity. Sections were then allowed to cool to room temperature, and were thereafter processed by the above immunocytochemistry procedure. As shown in Fig. 1, hsp72 detection was dramatically increased by this procedure, as was that of GFAP, whereas hsp27 detection was reduced. The already strong MAP2 signal was somewhat further enhanced (not shown), and this antigen retrieval method was therefore applied to the processing of all sections except those used for hsp27 detection. It is important to note that, whereas the antigen retrieval procedure employed here significantly enhanced hsp72 detection, it did not reproduce the dramatic shift in apparent distribution of hsp72 suggested in a preliminary report from this laboratory (Nowak et al., 1999).
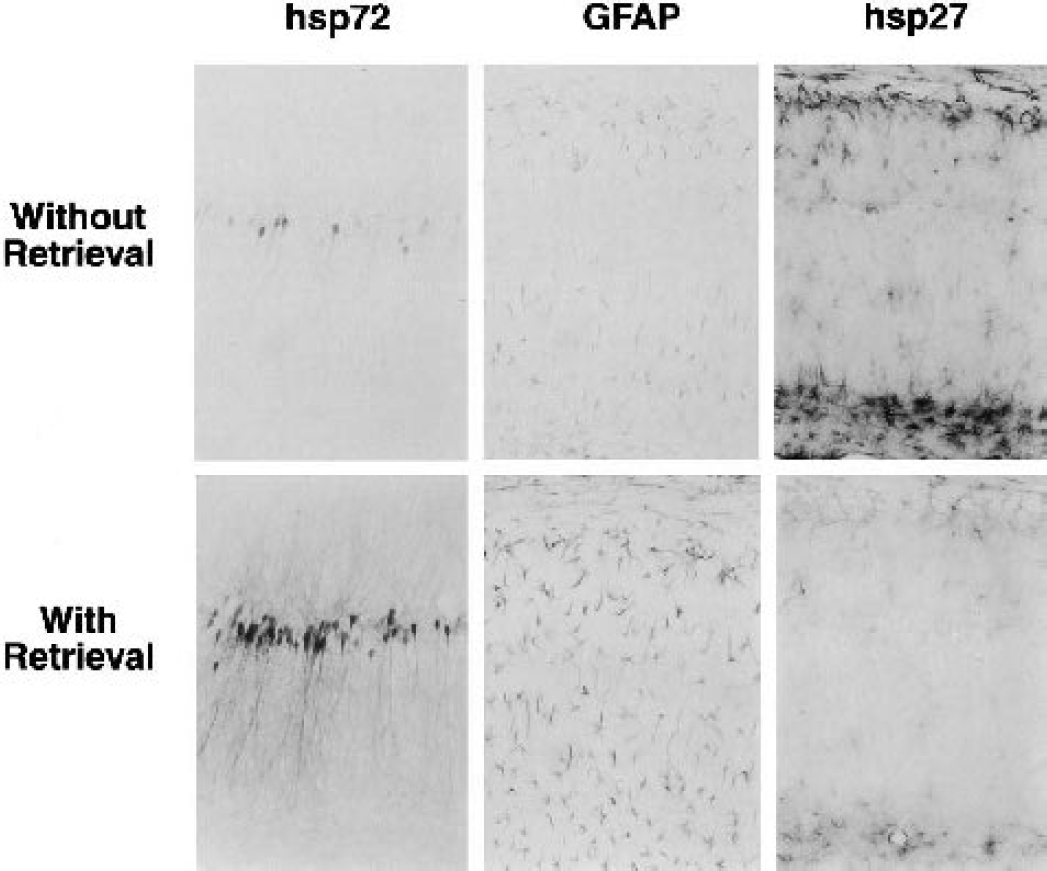
Effect of an antigen retrieval procedure on hsp72, glial fibrillary acidic protein (GFAP), and hsp27 detection. Gerbils were subjected to severe ischemic insults producing 6- to 8.5-minute depolarization followed by 4 days of recirculation, and vibratome sections were prepared and processed for immunocytochemistry with and without the antigen retrieval steps described in the text. Hsp72 and GFAP signals were notably enhanced by the retrieval procedure, whereas hsp27 detection was attenuated.
Statistical analyses
CA1 neuron counts were compared using analysis of variance and Scheffé F test implemented in StatView 5.0 (SAS Institute, Cary, NC, U.S.A.). Select immunocytochemistry scores were compared by χ-square analysis using the same program.
RESULTS
Depolarization thresholds for preconditioning and injury
Initial studies confirmed the reliability of ischemic depolarization as a predictor of hippocampal protection and injury, and characterized the preconditioning obtained with a 1-week interval between priming and test insults. These preliminary experiments verified a depolarization threshold of approximately 4 to 6 minutes for injury to naïve hippocampal CA1 neurons (Fig. 2), whereas an interval of 2- to 3.5-minute depolarization produced optimal ischemic tolerance induction. Hippocampi experiencing insults in the range chosen for standard test depolarizations in subsequent studies (6 to 8.5 minutes) showed robust protection after preconditioning, with a surviving population of 290 ± 19 cells/mm (n = 17) vs. 30 ± 18 cells/mm (n = 10) in naive hippocampi (P < 0.0001). Nevertheless, there was a small but statistically significant (10%) loss of CA1 neurons in optimally preconditioned hippocampi relative to the 321 ± 19 cells/mm (n = 11) in hippocampi experiencing the sham procedure or short insults of ≤3-minute depolarization (P = 0.0007).
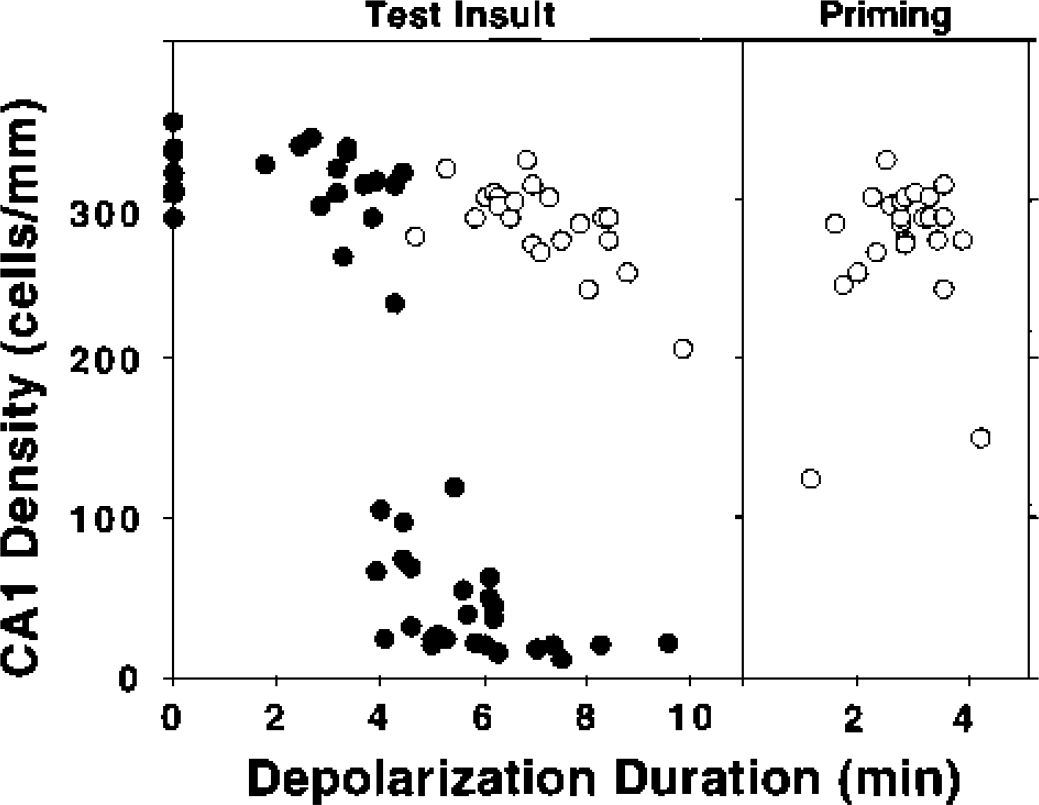
Depolarization thresholds for CA1 injury and tolerance induction in gerbil hippocampus.
Hsp72 expression
Hsp72 was present in ependymal cells but not in any hippocampal cell population in sham-operated animals (Fig. 3). After priming insults, hsp72 was moderately but consistently induced in dentate granule cells at 6 hours of recirculation, and this signal declined by 1 day. In contrast, CA1 pyramidal cells in approximately half of the cases expressed hsp72 at 2 and 4 days. In such positive hippocampi, expression was mainly localized in cell soma at 2 days, but became more intense and included dendritic processes at 4 days. Such expression was transient, consistently returning to basal levels by 7 days of recirculation. In the test insult group, hsp72 signal was faintly detected in dentate granule cells at 6 hours and the intensity peaked at 1 day. A strong hsp72 signal was frequently seen in CA3 at 2 days, but only scattered CA1 neurons showed staining in cell bodies and dendrites. The slight CA1 signal persisting at 4 days was characterized by a notable beaded appearance of the dendrites that was also reflected in MAP2 staining (not shown). Sparse staining was faintly detected in the few surviving CA1 neurons at 7 days. In tolerant animals, an otherwise severe insult induced no significant hsp72 expression at 6 hours of recirculation. By 1 day, hsp72 was diffusely expressed in dentate granule cells and CA1 neurons, indicating a somewhat more rapid accumulation in pyramidal cells than was seen after either priming or test insults alone. The signal increased at 2 and 4 days and dendritic staining in CA1 was observed, whereas signal in dentate granule cells gradually decreased, and essentially baseline expression was observed throughout hippocampus by 7 days.
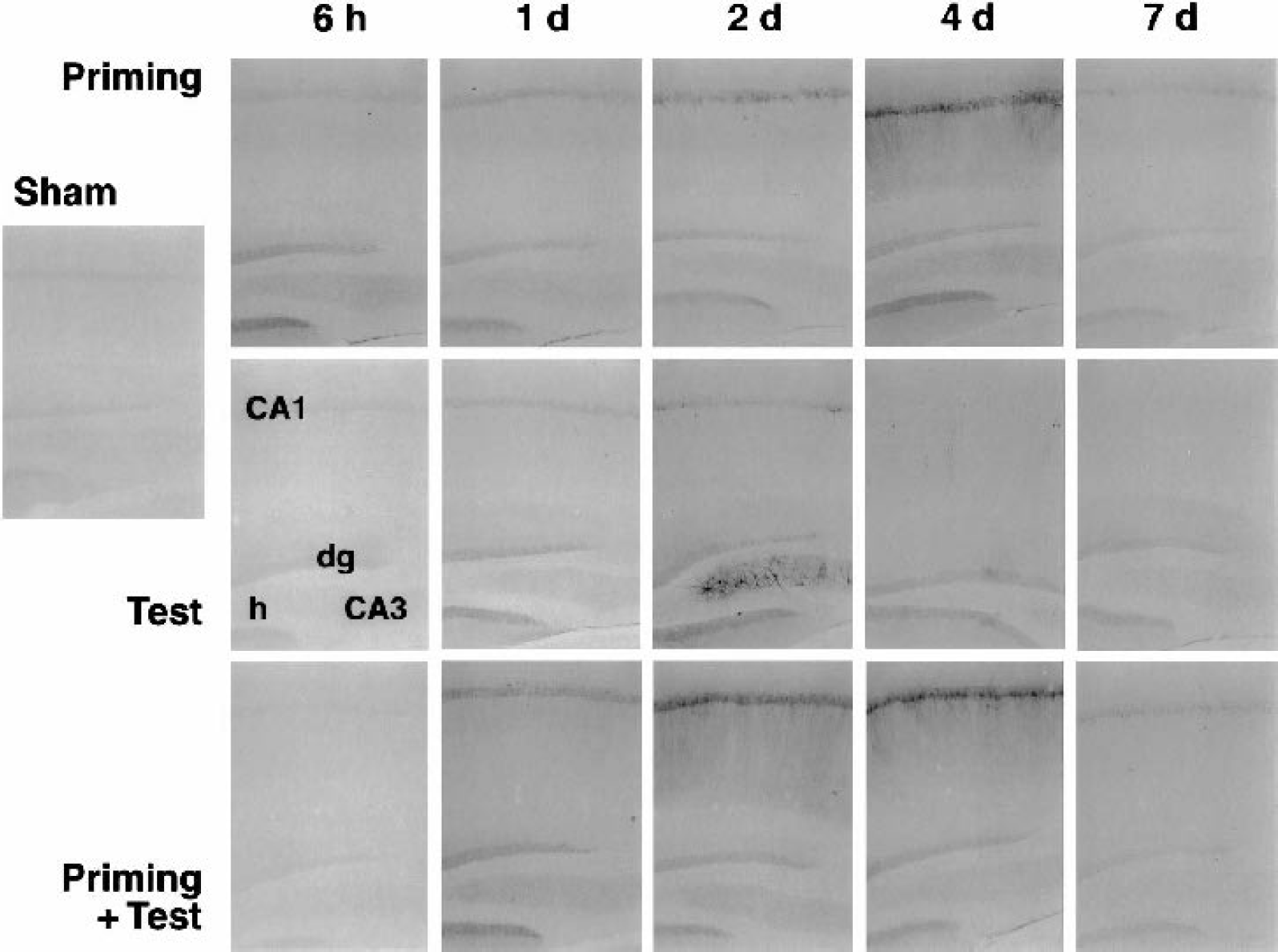
Distribution and time course of hsp72 expression in hippocampus after priming and test ischemic insults, and the effect of preconditioning. Gerbils were subjected to priming ischemia (2- to 3.5-minute depolarization), severe test ischemia (6- to 8.5-minute depolarization), or a combination of the two insults 1 week apart. Vibratome brain sections were prepared and processed for hsp72 immunocytochemistry with antigen retrieval at the time points indicated. Approximately half of the hippocampi exhibited moderate hsp72 expression in CA1 at 2 to 4 days after priming insults, returning to control levels by 7 days. Test insults in naive animals produced only a weak CA1 signal, with robust but transient CA3 expression in 50% of hippocampi at 2 days. The same insults to preconditioned hippocampi led to moderate, early hsp72 expression in dentate granule cells (dg) and consistent CA1 expression at 2 to 4 days. No appreciable signal was evident in dentate hilus (h) under these conditions.
Hsp27 expression
Basal hsp27 expression was absent from the hippocampus, apart from occasional profiles of large vessels of the hippocampal commissure in sham-operated animals (Fig. 4), other rare cells with one or two positive processes that were similarly distributed (not shown), and prominent staining of ependyma (Fig. 5). All animals showed prominent foci of astrocytic hsp27 expression at sites of cortex exposure, as well as along the electrode tracks, at postsurgery intervals of 1 day or longer (not shown), and these sites were avoided in evaluation of ischemia-induced changes. Priming insults increased hsp27 expression in dentate hilus at 1 day, increasing in intensity at 2 and 4 days, and persisting through 7 days, although 25% to 30% of hippocampi evaluated at each time point failed to show detectable expression. Double-labeling studies confirmed colocalization of hsp27 in GFAP-positive astrocytes (data not shown). No hsp27-positive glia were detected in CA1 after preconditioning. Persistent hsp27 expression was evident in dentate hilus in 100% of hippocampi after long test insults, increasing in intensity through 7 days. Astrocytes throughout CA1 showed pronounced hsp27 expression at 4 and 7 days that was coincident with delayed neuron loss. Preconditioned animals subjected to subsequent test insults exhibited residual hsp27 expression in dentate hilus at 6 hours, reflecting the response to the initial priming insult, and this signal did not change appreciably thereafter. As observed for naive animals, all preconditioned hippocampi subjected to the test insult exhibited some degree of hilar hsp27 expression. A weak signal was seen in the CA1 region of approximately half the hippocampi at 1 day and was evident in essentially all cases at 2 days, most prominently in stratum oriens. At 7 days, this signal was more closely associated with astrocytes in the pyramidal cell layer.
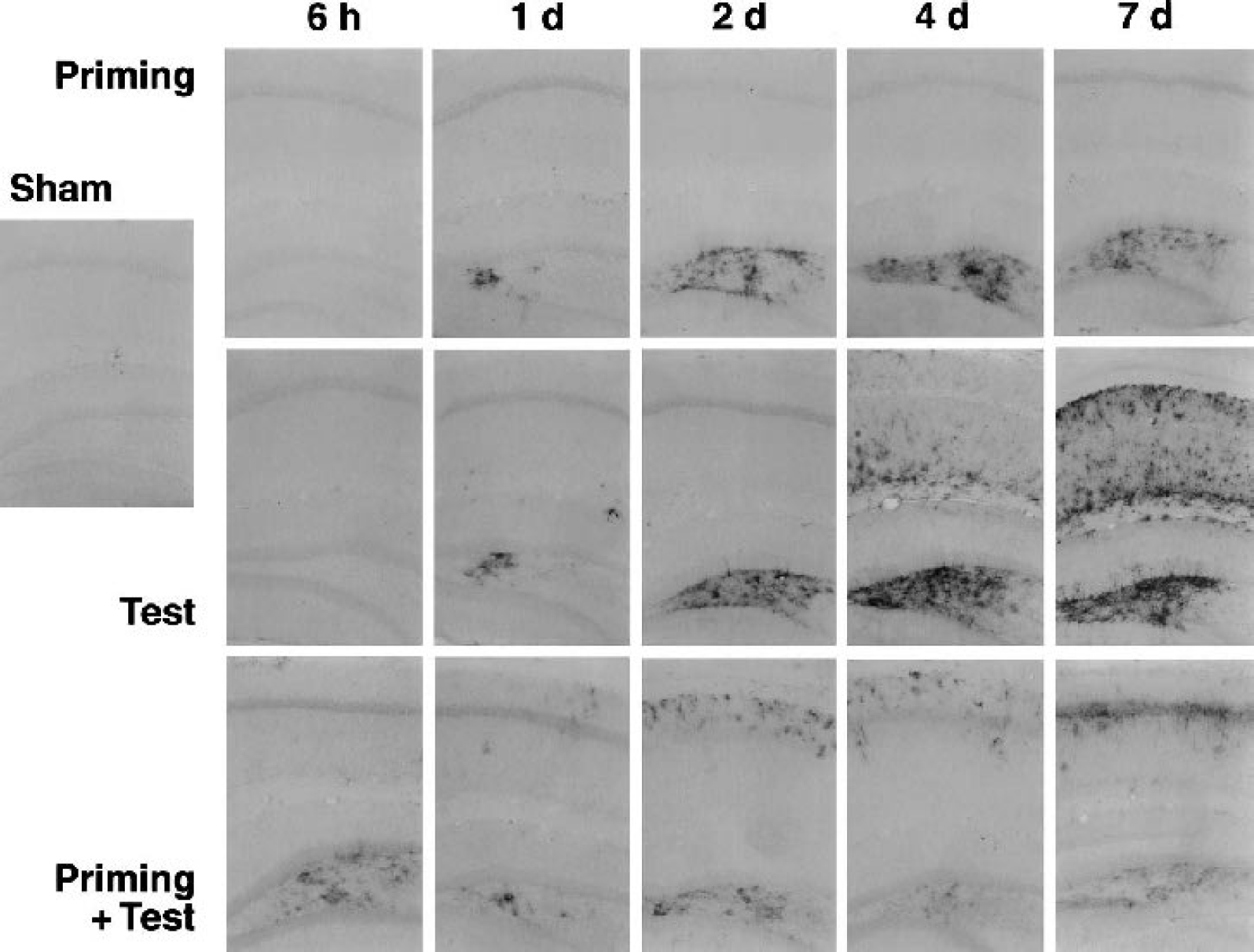
Distribution and time course of hsp27 expression in hippocampus after priming and test ischemic insults, and the effect of preconditioning. Gerbils were subjected to priming, test, or combined insults, and vibratome brain sections were prepared and processed for hsp27 immunocytochemistry at the time points indicated. Hsp27 expression was limited to dentate hilus after priming insults. Expression progressively increased during 1 to 4 days of recirculation and persisted at 7 days. Test insults to naive animals produced a similar time course of hilar hsp27 expression, more robust than seen after priming insults, and in addition showed marked CA1 involvement at 4 to 7 days. Test insults to preconditioned hippocampi did not produce any further change in hilar hsp27 expression beyond the residual signal from preconditioning, which persisted at an attenuated level. Scattered hsp27-positive profiles were evident in stratum oriens of CA1 during 1 to 4 days, with a more pronounced localization to the pyramidal layer at 7 days.
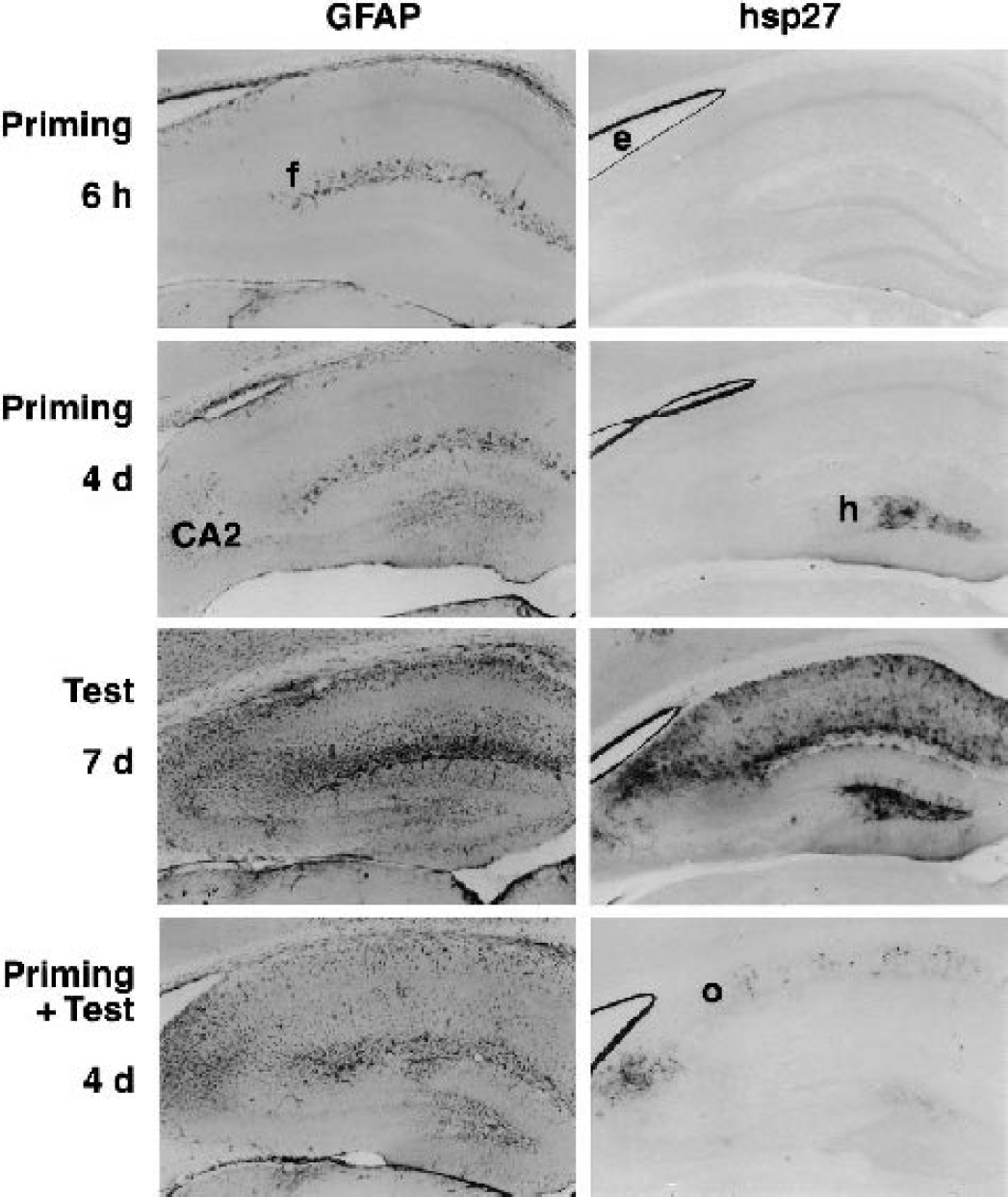
Comparison of glial fibrillary acidic protein (GFAP) and hsp27 distributions after ischemic insults. Nearby sections from representative hippocampi were processed in parallel for detection of GFAP and hsp27 immunoreactivities. Short priming insults produced no change in expression at 6 hours relative to sham-operated animals (data not shown), with GFAP-positive astrocytes detected periventricularly and at the hippocampal fissure (f), and hsp27 expression restricted to ependymal cells (e). At 4 days after priming insults, both signals increased in dentate hilus (h), whereas the CA2 region exhibited pronounced GFAP expression but relatively weak hsp27 staining. Severe test insults to naive animals resulted in markedly increased GFAP throughout the hippocampus at 7 days, primarily in CA2 and CA1 neuropil. Hsp27 was similarly distributed, but with a relatively more pronounced signal in dentate hilus. The same test insults to preconditioned hippocampus led to pronounced expression of both proteins in CA2, as well as in scattered cells in stratum oriens (o) of CA1, with a relatively weaker hsp27 signal in dentate hilus.
Colocalization of hsp27-positive astrocytes with regions of MAP2 loss
The distribution of hsp27 immunoreactive astrocytes shown above was clearly consistent with regions of known neuron injury in CA1 and dentate hilus after severe test insults, in which increases in GFAP immunoreactivity would also be expected. There was general agreement between the distribution of increased GFAP and hsp27 signals under all insult conditions, although relative responses differed somewhat among hippocampal regions (Fig. 5). For example, GFAP signal intensity was approximately equivalently increased in dentate hilus and CA2 4 days after priming insults, whereas the hsp27 response was much more pronounced in hilus. Hsp27 was also relatively more prominent than GFAP in dentate hilus 7 days after severe test insults. Preconditioned hippocampi showed enhanced GFAP signal throughout the CA1 region in response to a test insult, whereas hsp27 immunoreactivity was primarily evident in discrete foci preferentially localized in stratum oriens of CA1. However, both proteins exhibited prominent CA2 signals in such hippocampi. Hsp27 expression in these sections was generally associated with alterations in MAP2 immunoreactivity suggestive of subtle neuron injury or loss. As illustrated in Fig. 6, preconditioning insults resulted in appearance of hsp27 in dentate hilus within 1 day, and this was accompanied by moderate loss of MAP2-positive processes and accumulation of MAP2 in cell soma that remained evident through 7 days of recirculation. Likewise, the marked accumulation of GFAP- and hsp27-positive astrocytes in the CA2 region of preconditioned animals 2 and 4 days after test insults (noted previously) was associated with reproducible depletion of MAP2. Somewhat surprisingly, the decrease in the MAP2 signal in CA2 appeared to be transient, in that all five criterion hippocampi in which both proteins were evaluated 7 days after combined insults showed hsp27 staining in astrocytes without clear evidence of persistent MAP2 loss. A retrospective examination of the preconditioned hippocampi that provided the CA1 neuroprotection data shown in Fig. 2 revealed consistent eosinophilic CA2 neurons only after test insults of 7 minutes or longer, after which progressive CA1 loss also became evident. Scattered hsp27-positive astrocyte accumulation seen in CA1 after combined insults was associated with slight, persistnt MAP2 depletion through 7 days.
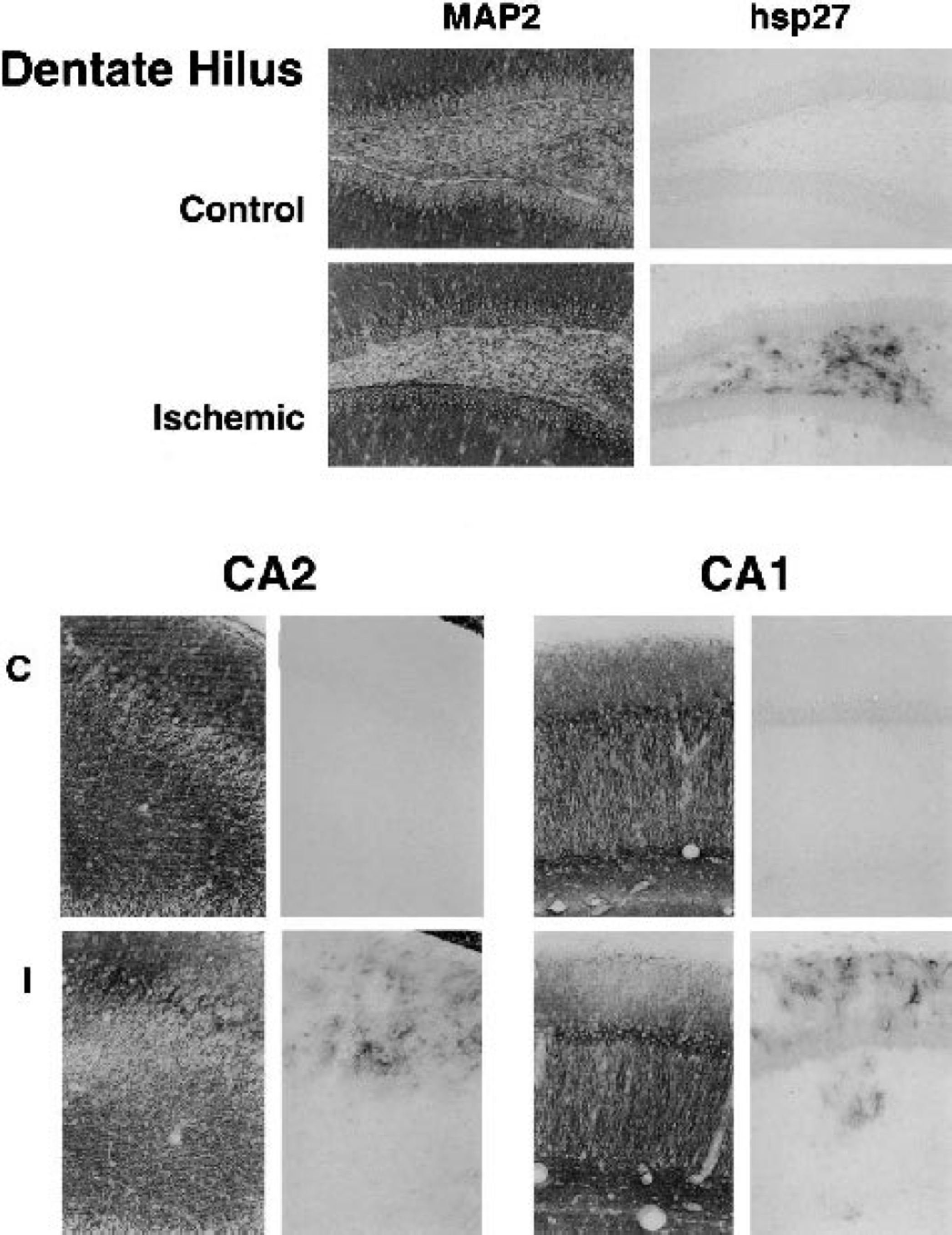
Hsp27 expression in regions of neuronal microtubule-associated protein 2 (MAP2) depletion. Nearby sections from representative hippocampi were processed in parallel for detection of MAP2 and hsp27 immunoreactivities. Hsp27 was absent from control tissue (C) except for ependyma, and was restricted to foci of MAP2 depletion after ischemia (I). Dentate hilus is shown 2 days after a priming depolarization, whereas CA2 and stratum oriens are illustrated for a preconditioned hippocampus 2 days after a subsequent test insult.
Depolarization thresholds for hsp72 and hsp27 responses
Grading of immunocytochemical signals provided a striking comparison of the depolarization thresholds for hsp72 expression in CA1 vs. hilar hsp27 induction. Although not intended as a primary endpoint of this study, there was sufficient heterogeneity in both preconditioning and test insults that such data could be extracted. The transient peak of hsp72 expression permitted meaningful evaluation only at the point of maximal expression at 4 days, whereas hsp27 data could be pooled from the 2-, 4-, and 7-days groups.
Hilar hsp27 expression was broadly dependent on insult severity, and a consistently robust signal was detected after depolarizations longer than 4 minutes (Fig. 7). However, the response to preconditioning depolarizations varied with the anatomical location of the section from a given animal. Undetectable or weak signal predominated in sections taken at a relatively rostral position at which the internal capsule was visible (<1.7 mm caudal to bregma). The χ-square comparison of rostral vs. caudal positions indicated a highly significant effect of section site on hsp27 grade in preconditioned hippocampi (P = 0.0006). Conversely, the level of sectioning did not impact signal detection after insults yielding depolarizations longer than 5 minutes. Additional sections were not available to formally examine the rostrocaudal distribution of hsp27 expression in the hippocampi of this study, having been used for other analyses. Accumulation of detectable hsp72 immunoreactivity in CA1 neurons was well correlated with insult severity even within the narrow range compatible with optimal tolerance induction, and was strongly evident only after depolarizations approaching 3 minutes (Fig. 8). In contrast, preconditioned hippocampi exhibited increased hsp72 expression after depolarizations of 6 to 8 minutes, over which range the hsp72 signal began to decline in naive hippocampi.
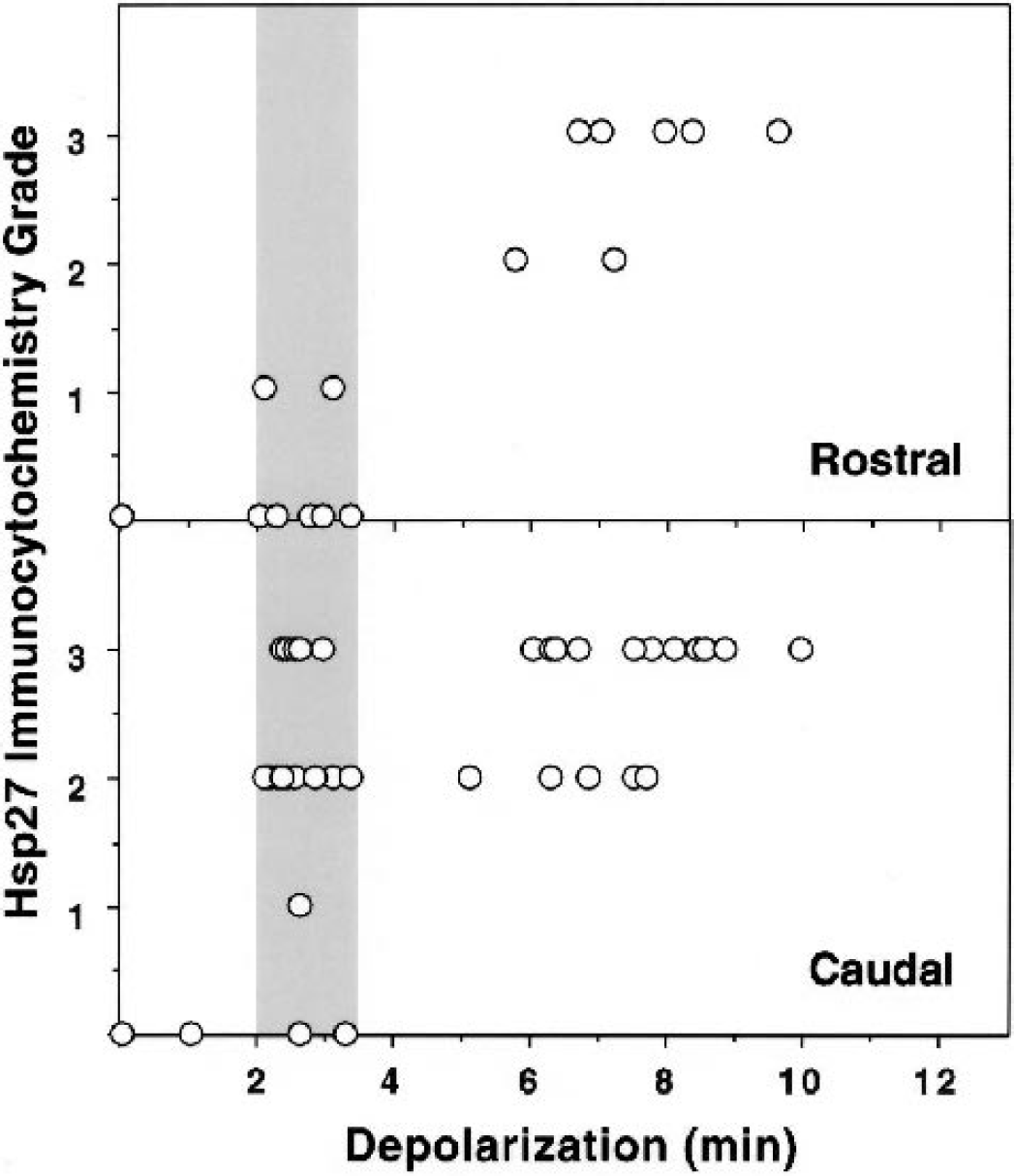
Regional variation in the depolarization dependence of hilar hsp27 expression. Immunocytochemical signals were detected and graded. The hilar hsp27 signal after preconditioning depolarizations (shaded bar) varied with the site at which sections were collected from a given animal. Samples collected at a slightly rostral level (upper panel) were predominantly negative after short preconditioning insults, whereas more caudal sections identified robust hilar hsp27 expression (lower panel). Such regional heterogeneity was not evident after the longer test depolarizations.
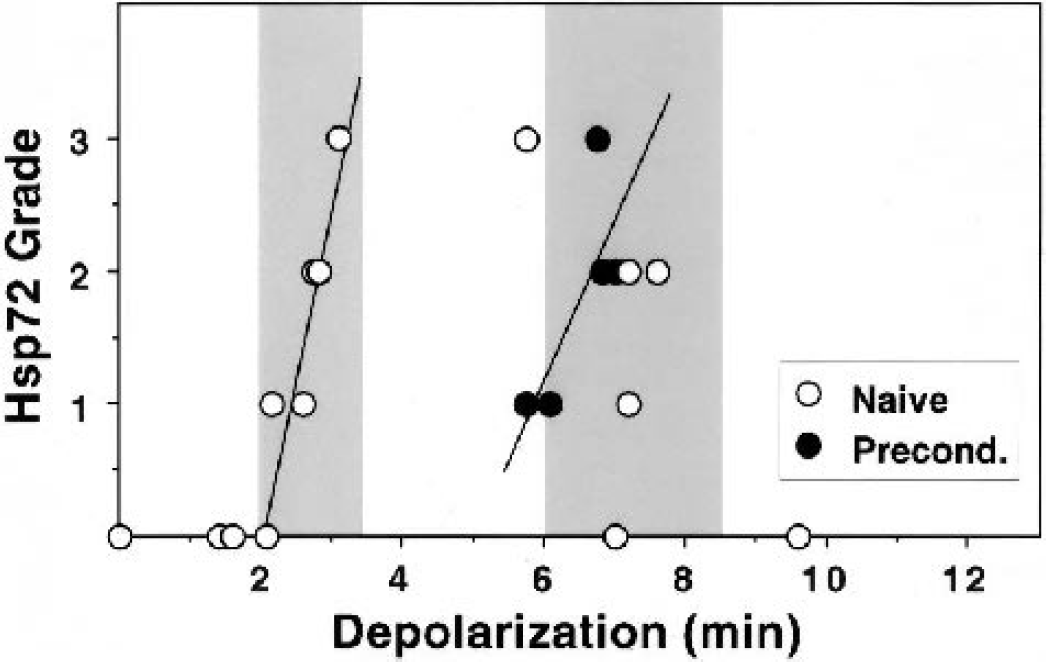
Depolarization thresholds for hsp72 expression in naive and preconditioned hippocampi. Immunocytochemical signals were detected and graded. CA1 hsp72 signal intensity in naive hippocampi progressively increased over the 2- to 3.5-minute depolarization range producing consistent preconditioning (left shaded region). Preconditioned hippocampi exhibited increasing hsp72 expression after insults longer than 6 minutes, in the range of standard test insults that reproducibly led to cell death and a decline in detectable hsp72 in naive hippocampi (right shaded region). Best fits to the rising phase of hsp72 expression in each group are provided for illustration.
DISCUSSION
These studies provide a detailed characterization of the time course and distribution of hsp27 and hsp72 protein expression in a highly controlled gerbil preconditioning model. The most striking observation is a robust accumulation of hsp27 in reactive astrocytes of dentate hilus beginning 1 day after preconditioning ischemia, in association with increased GFAP detection (Figs. 4 and 5). Together with persistent loss of MAP2 immunoreactivity in this region (Fig. 6), these results provide quantitative support for a previous suggestion that injury to vulnerable hilar interneurons occurs in the context of ischemic preconditioning (Matsuyama et al., 1993). No hsp27 expression was seen in protected CA1 neurons. The hsp72 results indicate that prior accumulation of this gene product is also not required for successful preconditioning (Figs. 3 and 8), complementing previous data at the mRNA level (Abe and Nowak 1996a, 2000, 2004). Rather, they indicate that preconditioned neurons exhibit an increase in the insult threshold for hsp72 induction.
Characteristics of the preconditioning model
The present results are in good agreement with a previous study in the gerbil (Abe and Nowak, 2004) with respect to both the insult threshold for CA1 loss and the range of depolarization durations consistent with optimal preconditioning (Fig. 2). An interval of 1 week between priming and test insults is longer than that used in most studies, and the shift in the depolarization threshold for injury in preconditioned hippocampi is less striking than seen with a more typical 2-day interval. However, although somewhat less robust, this model has the advantage of studying induced protection at an interval beyond the phase of preexisting hsp72 expression (Fig. 3). Although the responses observed in the combined insult group may not be those of maximally protected hippocampi, this choice clearly does not impact the determination of time course and depolarization threshold for a given response after single preconditioning insults.
Hsp27 expression and glial activation
The present hsp27 results are consistent with previous reports that expression is largely restricted to reactive astrocytes, as described in models of global ischemia (Kato et al., 1995, 1994b), focal ischemia (Imura et al., 1999; Kato et al., 1995), cortical lesions (Plumier et al., 1997a; Sanz et al., 2001), kainic acid administration (Kato et al., 1999), and repeated spreading depression (Plumier et al., 1997b). However, in contrast to one report of transiently increased CA1 neuron staining 1 day after global ischemia in a rat model (Kato et al., 1994b), we failed to detect hsp27 expression in major hippocampal neuron populations in any of our experimental groups. This may reflect subtle species differences in response, variations in relative insult severity, or perhaps antibody specificity or detection sensitivity. Antigen accessibility is a potential variable, but our attempts at antigen retrieval resulted in, if anything, a decrease in hsp27 detection.
Although a general correlation was noted between GFAP and hsp27 responses after severe insults in naive animals (Fig. 5), it is important to note that there could be significant dissociation of these signals. Priming insults alone resulted in a prominent GFAP response in CA2 that extended into adjacent CA3, but little hsp27 signal was detected in these regions after short insults. Test insults in preconditioned hippocampi resulted in diffusely increased GFAP immunoreactivity throughout CA1, whereas hsp27 was largely confined to stratum oriens. In all cases, hsp27 expression was restricted to regions that showed coincident attenuation of MAP2 signal in neuronal dendrites (GGFig. 6). Thus, whereas GFAP immunocytochemistry detected widespread glial activation in this and other preconditioning studies (Kato et al., 1994a), the present results indicate that hsp27 preferentially identifies an astrocyte subpopulation proximal to sites of neuron damage. This pattern is more restricted than described in an earlier study in a rat tolerance model, which showed hsp27-positive astrocytes diffusely distributed in hippocampus after a preconditioning insult (Kato et al., 1994b), perhaps indicative of a more severe priming insult in that study. Astrocytic hsp27 induction colocalized with regions of neuron damage after kainic acid-induced seizures (Plumier et al., 1996), and one focal ischemia study described most pronounced hsp27 expression in proximity to the infarct (Imura et al., 1999). However, widespread increases in hsp27 immunoreactivity in brain have been reported after focal ischemia (Kato et al., 1995), photothrombotic injury (Plumier et al., 1997a), or spreading depression (Plumier et al., 1997b), involving regions that do not show overt neuron injury. There appears to be a requirement for sustained intervals of propagated depolarizations to induce hsp27 expression, because short intervals of KCl application induce a limited hsp27 response to spreading depression (Plumier et al., 1997b). Seemingly parallel results have been reported for astrocyte nestin reexpression (Holmin et al., 2001; Lin et al., 1995).
The present results are in some respects also analogous to observations of selective vimentin upregulation in rat astrocytes in regions of neuron damage after global ischemia (Petito et al., 1990; Schmidt-Kastner et al., 1990) and focal cortical insults (Schiffer et al., 1986; Schroeter et al., 1995). Reactive astrocytes in many clinical neurodegenerative disorders show upregulation of vimentin (Yamada et al., 1992), hsp27, and another small stress protein, αB-crystallin (Head et al., 1993; Iwaki et al., 1993; Renkawek et al., 1994), although these can be dissociated under some conditions. Cortical lesions in early postnatal rat brain give rise to hsp27- and vimentin-positive astrocytes proximal to the site of damage, but result in a delayed appearance of hsp27-positive, vimentin-negative astrocytes in regions showing delayed neuron injury in thalamus (Sanz et al., 2001). Similarly, αB-crystallin was reported to show more limited induction than hsp27 after focal ischemia (Imura et al., 1999) and kainic acid administration (Kato et al., 1999), and was not found to increase after global ischemic insults that induced hsp27 (Kato et al., 1994b). Together the above results suggest that hsp27 and nestin expression may characterize a transition in glial activation, defining a subset of astrocytes showing increased GFAP, but not necessarily coupled to vimentin or αB-crystallin expression.
Hsp27 expression and hilar neuron injury
Sustained hsp27 expression and MAP2 depletion in dentate hilus after preconditioning insults are consistent with the well-known vulnerability of hilar interneurons to transient global ischemia (Johansen et al., 1987; Matsuyama et al., 1993; Sugimoto et al., 1993). It has been previously suggested that loss of somatostatin-immunoreactive hilar neurons after short priming insults could contribute to the tolerance mechanism (Matsuyama et al., 1993), and astrocyte and microglial responses consistent with injury to hilar neurons have also been reported after short insults (Kato et al., 1994a). However, another study using laminin staining suggested that hilar neurons, although not protected against subsequent insults, were not grossly injured by the priming ischemia (Sugimoto et al., 1993). Slight differences in insult severity related to details of priming and test conditions, such as postischemic temperature, could readily contribute to such discrepancies, and differences in tissue sampling also emerge as a potential contributor to apparent heterogeneity in hilar injury after short ischemic insults (Fig. 7). The extent of postischemic hilar lesions has not been formally characterized in the gerbil, but the present results are consistent with an early examination of insult thresholds in the rat that showed discrete localization of CA4 (hilar) damage to a single rostrocaudal level after 4-minute ischemia, expanding to the entire hippocampus after 10-minute insults (Smith et al., 1984). Although the weight of evidence indicates that localized hilar injury is a characteristic feature of preconditioning ischemia, there are strong arguments against a direct protective role of such lesions. Preconditioning clearly decays with time after a priming insult (Kato et al., 1991; Kirino et al., 1991; Kitagawa et al., 1991), yet can be reinduced long after such decay by a second preconditioning treatment (Chen et al., 1994). Nevertheless, it may yet be of interest to comprehensively examine the long-term plastic changes in hippocampal circuitry that occur after those hilar lesions that accompany ischemic preconditioning.
Hsp27 expression and reversible MAP2 depletion in protected hippocampal regions
Consistent hsp27 expression was seen in CA1 at 4 and 7 days after severe insults to naïve hippocampus (Fig. 4), coincident with the progression of neuron injury in this region. This was preceded at 2 days by a modest hsp27 signal in CA2 that became robust and contiguous with the CA1 response at later intervals (Fig. 5). However, of particular significance in the context of preconditioning is the evidence of subtle impacts on CA2 after priming insults alone and after test insults in preconditioned hippocampi. Foci of increased GFAP detection were evident in CA2 after priming insults (Fig. 5), showing only slight hsp27 expression at 4 days that was no longer evident at 7 days. There were no changes in MAP2 staining in CA2 neurons after preconditioning insults alone (data not shown). Subsequent test insults to preconditioned hippocampi resulted in consistent hsp27 expression in CA2 at intervals of 2 days or longer, accompanied by MAP2 depletion at 2 and 4 days (Fig. 6). Such observations are compatible with the relative vulnerability of CA2 and subiculum/medial CA1 after global ischemia, second only to that of dentate hilus in rats (Cizkova et al., 1996; Smith et al., 1984), mice (Yang et al., 2000), and gerbils (Hatakeyama et al., 1988; Iwai et al., 2000, 1993). It has been noted that CA2 and subiculum may constitute an anatomically continuous population of vulnerable neurons surrounding CA1 (Yoshimi et al., 1991). Previous studies have indicated a failure to protect this population after conventional ischemic preconditioning (Kirino et al., 1991; Nakagomi et al., 1993). However, the present results indicate transient MAP2 loss in CA2, with apparently normal staining in animals examined 7 days after repeated insults that nevertheless displayed hsp27 in this region. At this time point, eosinophilic CA2 neurons were prominent only in those preconditioned hippocampi experiencing test depolarizations of 7 minutes or longer (not shown), after which CA1 neurons were also less protected (Fig. 2). Because CA2 neurons are spared in the present model under the same conditions that maximally preserve CA1 neurons, hsp27-positive glia and transient MAP2 reduction appear to characterize a reversible injury to CA2 neurons after optimized preconditioning, consistent with the “reactive change” noted after single insults by histological and immunocytochemical methods (Hatakeyama et al., 1988; Kirino and Sano 1984; Yamashita et al., 2003; Yoshimi et al., 1991).
Preconditioned hippocampi in rare cases showed diffusely reduced MAP2 and scattered hsp27-positive astrocytes as expected immunocytochemical correlates to partial loss of CA1 neurons. However, more typical was MAP2 loss and hsp27 expression restricted to CA1 stratum oriens (Figs. 4–6), without evident involvement of the soma or apical dendrites, indicating that overt pyramidal cell death did not underlie these changes. Interestingly, early fodrin proteolysis has been described in stratum oriens in the gerbil after ischemia (Yokota et al., 1995), indicative of regional cytoskeletal lability preceding neurodegeneration. Reversible alterations in dendritic morphology, including changes in MAP2 distribution, have been reported in cultured neurons after sublethal excitatory amino acid exposure (Bigot et al., 1991; Park et al., 1996). There is evidence for loss of MAP2 immunoreactivity after hypoxia/ischemia in immature rat brain before the appearance of histologic damage (Gilland et al., 1998), and the possibility of remarkable recovery of MAP2 staining after such insults has been suggested (Ota et al., 1997). Many proteins such as MAP2 are quite labile in postmortem gerbil hippocampus, and early immunocytochemical changes in CA1 have been reported under some conditions (Matesic and Lin 1994; Yanagihara et al., 1990) but have only been observed at later intervals in other studies (Yoshimi et al., 1991; Saito N, Nowak TS Jr, unpublished observation, 1991). This appears to reflect sites of local protease activation during tissue processing, potentially amplifying or even falsely reporting the in vivo condition. Other studies in the rat indicated that such changes occurred more slowly if at all (Irving et al., 1997; Tomioka et al., 1992; Vanicky et al., 1995), and it has since been shown that early postmortem hippocampal proteolysis is more prominent in the gerbil than in the rat or mouse (Kitagawa et al., 1999). Although the present results suggest that astrocytic hsp27 expression is potentially associated with reversible stages of neuronal injury in preconditioned CA1, more detailed studies are needed to adequately define the underlying responses in these neurons.
Hsp72 expression and CA1 vulnerability
The present results provide further evidence against a role for prior accumulation of the 70-kd stress protein, hsp72, in ischemic preconditioning. We have previously noted that only half of the hippocampi experiencing depolarizations in the range producing optimal tolerance induction showed detectable hsp72 mRNA accumulation in CA1 neurons during early recirculation (Abe and Nowak, 2004). This study likewise shows that although hsp72 immunoreactivity could be detected in CA1 after the longer priming insults (Figs. 3 and 8), this was not observed after shorter depolarizations in the range inducing maximal tolerance. Furthermore, hsp72 was no longer detectable at 7 days after priming insults (Fig. 3), at which point substantial tolerance could still be observed (Fig. 2), reinforcing the temporal dissociation between hsp72 expression and protection noted in earlier studies (Kirino et al., 1991).
There was some evidence of more rapid hsp72 expression in CA1 of tolerant animals at 1 day after the severe test insult (Fig. 3), consistent with reports of accelerated hsp72 mRNA induction in other tolerance studies (Aoki et al., 1993; Kanemitsu et al., 1994). The strong hsp72 signal in CA1 at 2 and 4 days of recirculation presumably reflects in large part the increased translational capacity of the protected neurons as detected by in vivo labeling (Nakagomi et al., 1993; Nishi et al., 1993) and in tissue slices (Paschen and Mies, 1999). It remains unclear whether this apparent potentiation of hsp72 expression in tolerant neurons plays a direct role in cell survival or rather reflects the fact of such protection. For example, an abbreviated time course of hsp72 protein expression was reported in a kainic acid preconditioning model (Blondeau et al., 2000), suggestive of a milder insult. The present results indicate that the depolarization threshold for hsp72 induction is increased in preconditioned CA1 (Fig. 8). Although the number of samples supporting this incidental finding is small, the predictive power of the current model lends considerable strength to the observation. There was also no detectable hsp72 expression in CA3 neurons under these conditions (Fig. 3). It would therefore appear that cellular stress, as indicated by hsp72 expression, is reduced throughout hippocampus, and presumably throughout the brain (Kitagawa et al., 1991), strongly suggesting that preconditioned neurons experience a reduction in insult severity at a step before activation of the signals leading to hsp72 induction. Recent results indicate attenuated levels of protein aggregates after preconditioning (Liu et al., 2003), perhaps reflecting in part reduced protein denaturation. Together with observations of reduced calcium entry (Ohta et al., 1996; Shimazaki et al., 1998) and reduced oxidative damage (Baek et al., 2000) during subsequent ischemic insults, there emerges a pattern of insult attenuation at an early step in the ischemic injury cascade after preconditioning.