
Abstract
The α-amino-3-hydroxy-5-methyl-4-isoxazole (AMPA) receptor antagonist, 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline (NBQX), offers protection to hippocampal CA1 pyramidal cells after short episodes of transient cerebral ischemia. Besides CA1 pyramidal cells, neurons containing somatostatin (SS) and located in the dentate hilus of the hippocampal formation are lost after cerebral ischemia. We studied the protective effects of NBQX on SS neurons in the hilus and on hippocampal CA1 pyramidal cells following 8, 10, or 12 min of four-vessel occlusion ischemia during systemic hypotension. NBQX was administered 3 × 30 mg/kg at 0, 10, and 25 after induction of ischemia or sham, and all rats survived for 7 days. NBQX given to control rats without ischemia had no influence on number or morphology of hilar SS neurons and CA1 pyramidal cells. After 8 min of ischemia, NBQX prevented loss of hilar SS neurons. After 10 and 12 min of ischemia, NBQX had no significant effects on loss of SS neurons in the dentate hilus. However, in all ischemic groups, NBQX significantly reduced loss of CA1 pyramidal cells as compared to control rats. This neuroprotective effect decreased gradually and significantly as the time of ischemia increased. Our results support the observation that SS neurons in hilus are among the most ischemia-vulnerable neurons in the brain. We found that administration of NBQX in generally accepted dosages can protect the rapidly dying SS neurons in hilus from only brief episodes of ischemia.
Disturbed glutamatergic neurotransmission is one important mechanism involved in ischemic cell death of several neuron types (Benveniste et al., 1984; Andiné et al., 1991). Thus, removal of the glutamatergic input to the vulnerable CA1 pyramidal neurons has a protective effect (Wieloch et al., 1985; Johansen et al., 1986; Benveniste et al., 1989). Accordingly, it has been shown that glutamate antagonists at the α-amino-3-hydroxy-5-methyl-4-isoxazole (AMPA) receptor have protective capabilities against ischemic neuron loss in hippocampus CA1, striatum, neocortex, and cerebellar cortex (Sheardown et al., 1990; Nellgård and Wieloch, 1992; Balchen and Diemer, 1992; Diemer et al., 1990, 1992). In contrast to the delayed ischemic cell death seen in CA1, irreversible damage (eosinophilia) of hilar neurons containing somatostatin (SS) develops rapidly (Johansen et al., 1987). Since it is unknown whether AMPA receptor-mediated mechanisms are involved in ischemic cell death of this particular cell type, we studied the neuroprotective effects of an AMPA antagonist, 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo(F)quinoxaline (NBQX) on hilar SS neurons, and compared results with its effects on hippocampal CA1 pyramidal cells.
MATERIALS AND METHODS
The experimental protocol was approved by the Danish State Animal Inspectorate. Adult male Wistar rats (350–450g) had their vertebral arteries electrocauterized (Pulsinelli and Brierley, 1979) under methohexital anesthesia (50 mg/kg i.p.). They were then fasted for 24 h with free access to water before they were reanesthetized, intubated, and ventilated with 1.2–1.6% halothane in a gas mixture of O2:N2O (vol/vol) in the scale 1:2. A femoral artery was cannulated, connected with a syringe, and the following parameters measured: pH, blood gases, MABP, and plasma glucose (PG). The carotid arteries were exposed and halothane was disconnected 2 min before the carotids were ligated for 8, 10, or 12 min, during hypotension established by withdrawing blood (Benveniste et al., 1989). Rectal and scalp (temporal muscle) temperatures were monitored and both regulated by heating lamps. Ischemia was terminated by removing the carotid ligatures and reinfusing the withdrawn blood, supplemented with 1–2 ml saline, until MABP of >100 mm Hg was reached. Both rectal and scalp temperatures were controlled (setting 37°C) during the first hour after ischemia before the animals were moved to their normal 22°C stable facilities in cages. Nonischemic sham-operated control rats were injected three times (0, 10, and 25 min) with 1 ml isotonic glucose i.p. (vehicle) or 30 mg/kg of NBQX (NOVO Nordisk CNS Division, Måløv, Denmark) dissolved in isotonic glucose. Ischemic rats were injected i.p. with vehicle or NBQX (30 mg/kg) three times (0, 10, and 25 min) after the carotid ligatures were released.
All rats were killed via deep halothane anesthesia by perfusion fixation with 4% paraformaldehyde in 0.15 M phosphate buffer at 7 days after the ischemia or sham operation. Brains were removed from the skulls and subsequently postfixed for 4 h in the same fixative. Then, 50 μm thick coronal sections of the dorsal hippocampus, between 2.4 and 2.6 mm caudally from the bregma, were cut on a Lancer-1000 vibratome (Technical Products International, St. Louis, MO, U.S.A.) and immunocytochemically stained according to previously described standard procedures (Johansen et al., 1987). We used SS antibodies (diluted 1:1,000) raised against sequence 5–10 of SS-14 (Prof. J. J. Holst, Department of Medical Physiology, University of Copenhagen). Immunosections were counterstained with hematoxylin and eosin (H&E). SS immunopositive neurons were counted in the dentate hilus at 250× magnification by light microscopy. Results were calculated per 480 μm grid length of the dentate granule cell layer. The remaining part of the brains were embedded in paraffin for subsequent H&E staining. Coronal sections, 10 μm thick, were cut and collected from 2.7 mm behind bregma and more caudally. Finally, CA1 pyramidal cells were counted in the light microscope per 300 μm grid length placed in the medial, intermediate, and lateral parts of the CA1 stratum pyramidale of the ipsi- and contralateral dorsal hippocampus at 2.70–2.75 mm caudally from the bregma.
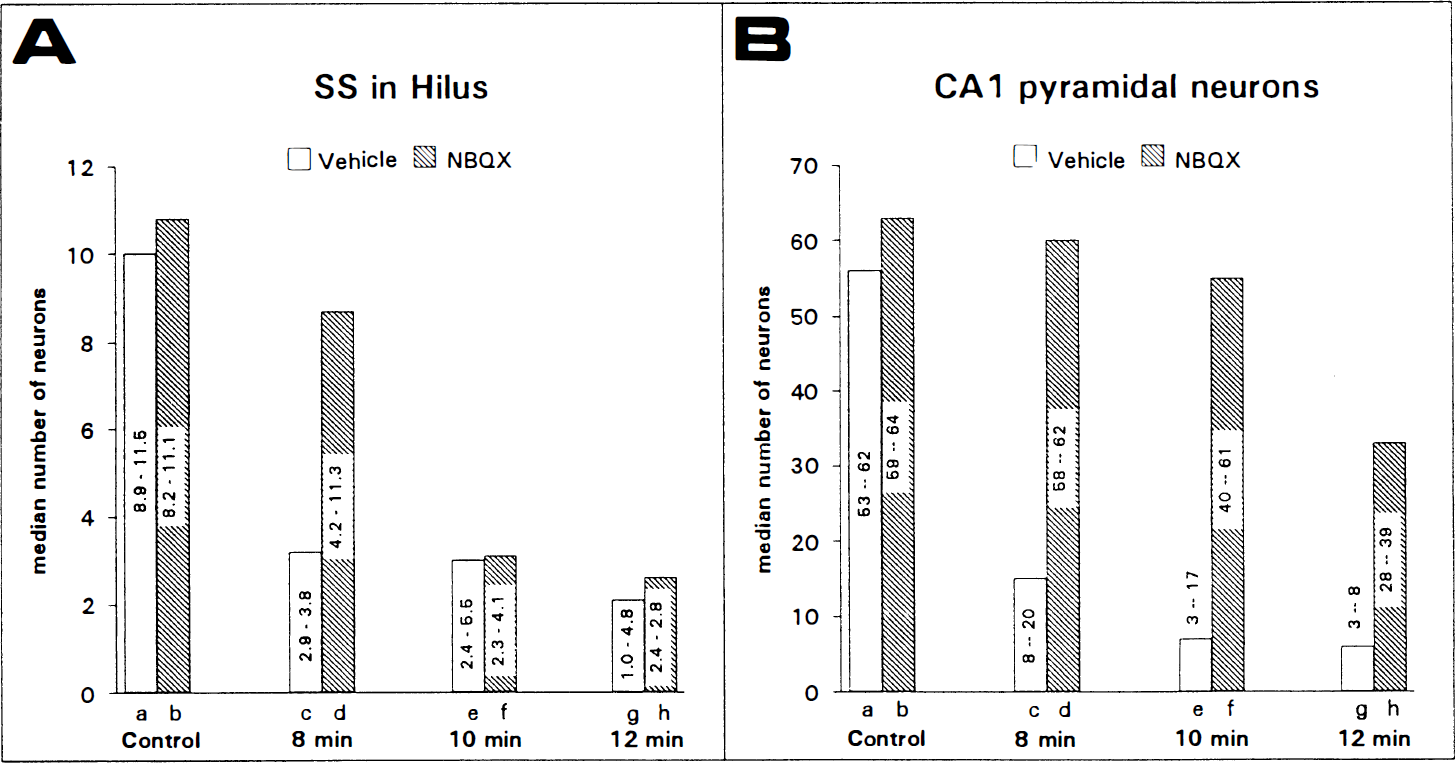
Histologic values from rats who received vehicle were compared to NBQX-treated rats by Mann–Whitney statistics. Comparisons of vehicle versus vehicle treatment or NBQX versus NBQX treatment between groups with different ischemic insults (i.e., controls, 8, 10, or 12 min) were performed with the Kruskal–Wallis one-way analysis of variance (ANOVA) by ranks. Comparisons performed are indicated in Fig. 1. Statistical significance was accepted for p < 0.05.
RESULTS
Physiologic values are listed in Table 1. These were examined qualitatively and no substantial differences were observed.
Physiological variables
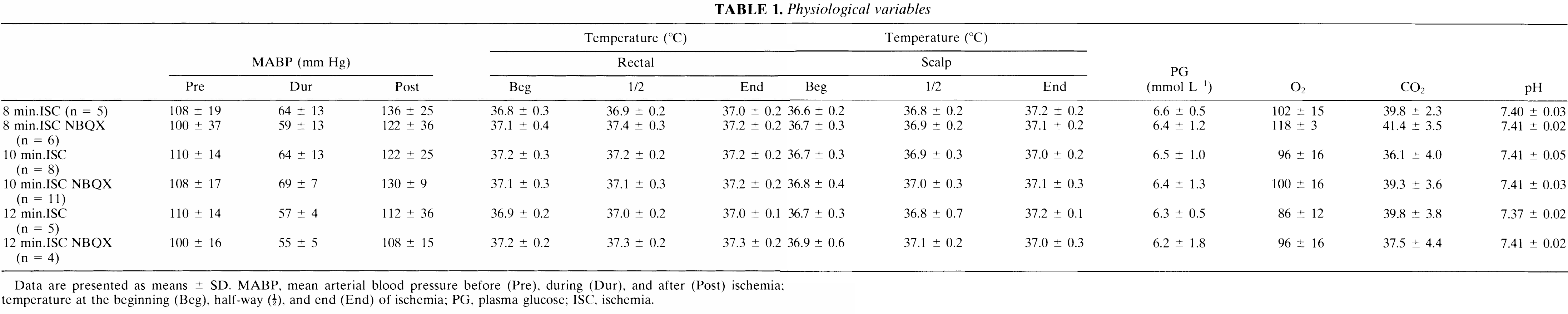
Data are presented as means ± SD. MABP, mean arterial blood pressure before (Pre), during (Dur), and after (Post) ischemia; temperature at the beginning (Beg), half-way (½), and end (End) of ischemia; PG, plasma glucose; ISC, ischemia.
SS neurons in control and ischemic animals
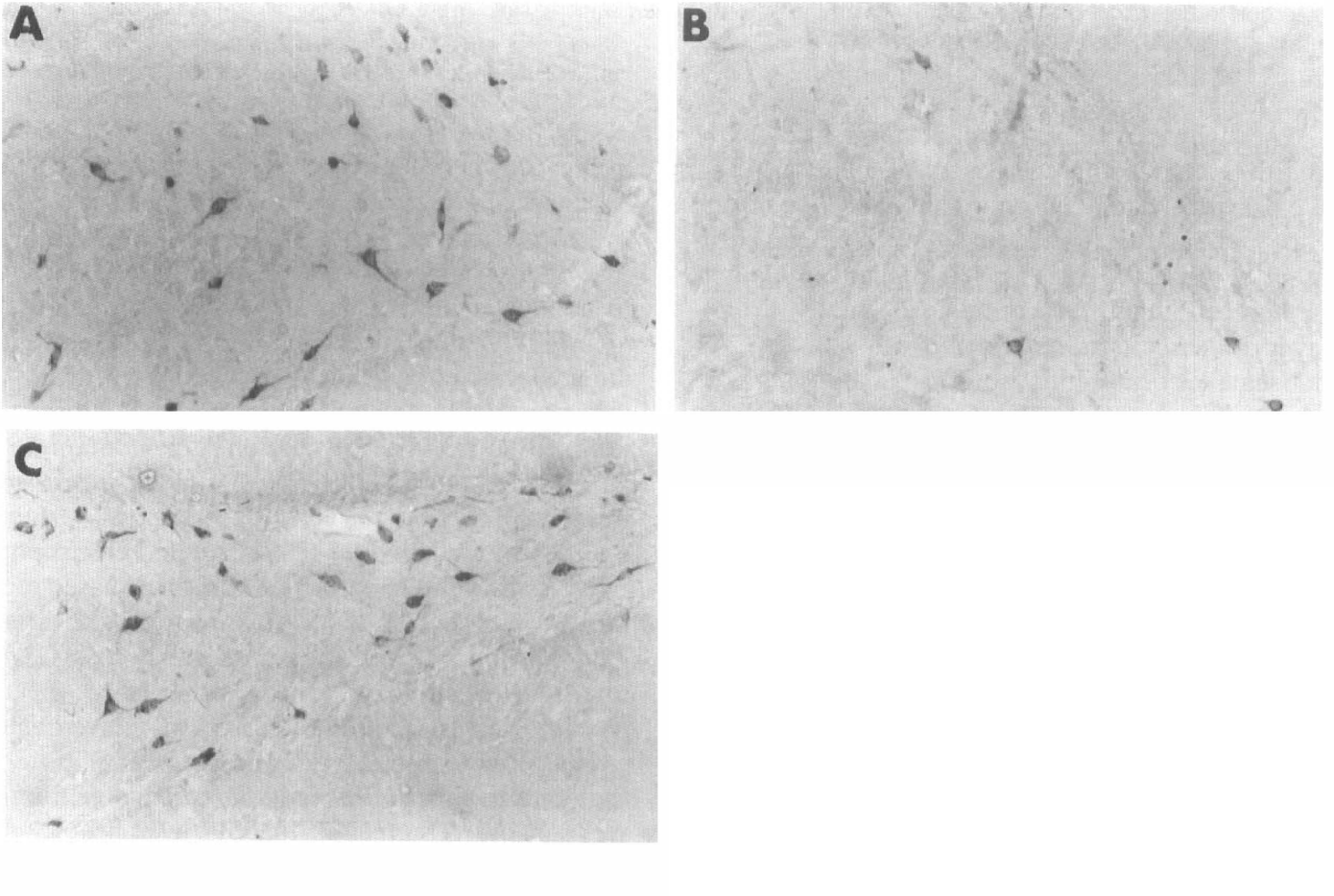
Systemic NBQX administration in control animals without ischemia had no influence on morphology (Fig. 2A) as compared to control animals injected with vehicle. Cell counts in these two groups showed no statistical differences in numbers of SS neurons in the dentate hilus (Fig. 1A).
In rats injected with vehicle and subjected to 8, 10, or 12 min of ischemia, there was a significant loss of SS neurons in hilus, as compared to vehicle-injected rats without ischemia (Fig. 1A).
After 8 min of ischemia, NBQX prevented loss of SS neurons in hilus (Figs. 2C, 1A), but after 10 and 12 min of ischemia, NBQX had no significant effects on the loss of SS neurons in hilus (Figs. 2B, 1A).
CA1 pyramidal cells in control and ischemic animals
Control rats who received either vehicle or NBQX showed no signs of neuropathological changes, e.g., eosinophilic neurons were not seen, and cell counts of CA1 pyramidal cells showed no significant differences in numbers between these groups (Fig. 1B).
In ischemic rats treated with vehicle, CA1 pyramidal cell loss was 75–90%, but statistical analysis (Fig. 1B) showed no significant differences between loss in the three groups. Loss of pyramidal cells were seen throughout CA1 to the very border of CA3.
In ischemic rats treated with NBQX, CA1 cell counts and statistical analysis in rats subjected to 8 and 10 min of ischemia demonstrated total protection when compared to NBQX-injected control rats (Fig. 1B), although a very small number of eosinophilic neurons could be observed in ischemic animals. Moreover, there was only partial protection against CA1 pyramidal cell loss in rats subjected to 12 min of ischemia (Fig. 1B). Also, statistical analysis showed a significant difference between loss in rats subjected to ischemia of 8 and 10 min, and 10 and 12 min, respectively (Fig. 1B). Again, eosinophilic neurons could be seen in all three groups. Ischemic damage to the CA1 pyramidal cells after NBQX treatment was randomly scattered throughout the entire CA1.
DISCUSSION
We have investigated the neuroprotective effects of NBQX administered systemically during the immediate reflow period (<25 min) after 8, 10, or 12 min of cerebral ischemia. Two types of vulnerable neurons in the rat hippocampal formation were studied: hilar neurons containing SS and CA1 pyramidal cells. After 8, but not after 10 or 12 min of ischemia, NBQX prevented loss of hilar SS neurons. In agreement with previous reports (Sheardown et al., 1990; Nellgård and Wieloch, 1992; Diemer et al., 1990, 1992), we found that NBQX ameliorated ischemic damage to CA1 pyramidal cells in all ischemic groups. The CA1 neuroprotective effect decreased gradually as the time of ischemia increased.
We previously demonstrated that hilar SS neurons are lost within the first 2 days after ischemia (Johansen et al., 1987, 1993). We called this rapid ischemic cell death in contrast to the 3–4 days delayed ischemic pyramidal cell death seen in CA1 and because eosinophilic neurons in hilus can be seen as early as 3 h after ischemia (Johansen, unpublished data). Furthermore, we observed that very short episodes of ischemia damages hilar neurons without any damage to the CA1 pyramidal cells. This is in agreement with findings by Matsuyama et al. (1993), who concluded that hilar SS neurons are more vulnerable to ischemia than are CA1 pyramidal cells. Indeed, ischemic damage to hilar neurons has been described (Ito et al., 1975). Recently, it has been suggested that far more neuron types than just SS neurons and mossy cells in hilus are damaged (Hsu and Buzsáki, 1993). We cannot directly compare our results with those of the latter study, but in SS immunosections counterstained with acid fuchsin (which marks ischemic eosinophilia), eosinophilic neurons without SS immunostaining were few in number.
The nature of the SS neurons in the dentate hilus is controversial. Kosaka et al. (1988) found a high degree of neuronal colocalization of SS and glutamate decarboxylase in hilus, whereas Sloviter and Nilaver (1987) concluded that hilar SS neurons belong to a neuron population not containing γ-aminobutyric acid (GABA). The latter view is indirectly supported by several observations. Using an epilepsy model, Sloviter (1991) studied electrophysiology in the dentate gyrus and found decreased recurrent granule cell inhibition associated with loss of hilar SS neurons and mossy cells, whereas GABA interneurons survived. On the other hand, electrophysiological recordings after ischemia have demonstrated increased recurrent granule cell inhibition in the dentate gyrus (Chang et al., 1989). Accordingly, we (Johansen et al., 1987, 1989b) found ischemic loss of the same neuron types as in epilepsy as well as survival of the GABA interneurons (Johansen et al., 1989a). However, in ischemia, mossy cells are lost only following prolonged insults (>30 min) (Johansen, unpublished data).
Both in experimental epilepsy (Sloviter, 1991) and ischemia (Johansen et al., 1987) SS neurons are lost, whereas GABA interneurons survive the insult. This suggests that hilar SS neurons do not contain GABA. Therefore, hilar SS neurons may be excitatory neurons. This is supported by the observation that AMPA binding increases postischemia in the outer molecular layer of the dentate gyrus (Diemer et al., 1987), which is deprived of axons after ischemic loss of hilar SS neurons (Bakst et al., 1986). If we consider that the SS neurons in hilus reinforce granule cell excitation through feedback excitation, then increased recurrent granule cell inhibition after ischemia (Chang et al., 1989) may simply follow loss of SS neurons and their feedback excitation.
Increases in intracellular calcium concentrations is suggested to play a key role in processes leading to ischemic cell death (Siesjö, 1981, 1988; Siesjö and Bengtsson, 1989). The AMPA receptor mediates fast excitatory transmission by increasing sodium and potassium ion fluxes (Verdoorn et al., 1991) and, thereby, stimulating calcium influx through activation of voltage-dependent calcium channels. After ischemia, stimulation of the AMPA receptor may result in an increased influx of calcium from the extracellular space. The AMPA receptor in itself may be or become permeable to calcium (Sommer et al., 1991) and/or ischemia may potentiate processes opening voltage-operated calcium channels in the cells. Stimulation of at least one type of metabotropic glutamate receptor can activate inositol triphosphate (Masu et al., 1991), thereby releasing intracellular stores of calcium. Ischemia may potentiate such receptor-coupled processes and mediate increased release of calcium from intracellular stores in vulnerable cells. Furthermore, stimulation of the metabotropic glutamate receptor in concert with the AMPA receptor causes release of arachidonate (Dumuis et al., 1990), which will potentiate NMDA receptor currents and amplify increases in the intracellular calcium concentrations caused by glutamate (Miller et al., 1992).
It is also possible that during and/or after ischemia, vulnerable cells are less capable of handling increased (Andiné et al., 1991) or even normal influxes of ions, especially divalent cations like calcium and zinc. In this connection, we speculate (Johansen et al., 1993) that hilar SS neurons may be very vulnerable to ischemia since they are surrounded by the zinc-rich mossy fiber boutons from which zinc, together with glutamate, is released during synaptic transmission (Charton et al., 1985).
Besides increase in intracellular cation levels, ischemic damage through receptor-operated mechanisms may involve changed activities of protein kinase C, phospholipase A2, arachidonate, and production of free radicals (Siesjö, 1981). Recently, it was suggested that ischemic disturbances in AMPA receptor activation might lead to detrimental changes in gene expression (Zafra et al., 1990). Whatever the ischemic process involves, it is likely to terminate with crucial increases in intracellular calcium concentrations leading to cell death in vulnerable neurons (Siesjö and Bengtsson, 1989).
A large number of studies, including those on deafferentation and pharmacological treatment, have permitted the conclusion that at least some forms of regional ischemic brain injury are convincingly mediated by excitotoxic mechanisms and altered calcium homeostasis. Lately, interest has been focused on the sedative and hypothermic effects of pharmacological treatment of ischemic brain damage. It has been demonstrated that brain temperatures at 30– 33°C during and/or after ischemia confer partial protection against brain damage. Ginsberg and coworkers (1993) have pointed out, in their review, that the first hour of postischemic recirculation is of particular critical importance in hypothermic protection of ischemic damage. For example, hypothermia reduces glutamate transmitter release, and the enzyme calcium/calmodulin-dependent protein kinase II, which is responsible for signal transduction linking intracellular increases in free calcium ion to regulation of protein phosphorylation, is inhibited by normothermic ischemia, but unaffected following hypothermic ischemia.
In our study, we kept the animals normothermic during the first postischemic hour and then transferred them in cages to their normal environment at room temperature (22°C).
The dosages of NBQX used kept the animals hypoactive for the first 3–4 h postischemia, but ischemic rats without NBQX treatment were also hypoactive in this period. Thus, examination of the significance of postischemic temperature changes in the period preceding cell death is now being carried out. In future studies, definitions of ischemic models may include long-term postischemic regulation of brain temperature. Thus, careful long-term regulation of brain temperature will further define the pathomechanisms with which one is dealing in pharmacological treatment of ischemic brain damage. We suggest that our NBQX treatment of ischemic damage in hippocampus primarily protected hilar SS neurons through mechanisms blocking glutamatergic neurotransmission immediately after ischemia. At the present time, we cannot exclude that synergistic protective effects could have been due to the influence of NBQX on brain temperatures after the first postischemic hour. From our study, we conclude that NBQX can protect SS neurons in the hilus from brief episodes of transient cerebral ischemia.
Footnotes
Acknowledgment:
The authors wish to thank Lisbeth Thatt for excellent technical assistance and Torben Balchen for critical evaluation of statistics.