
Abstract
There has been considerable interest in the use of thrombin inhibitors to reduce the occurrence of stroke or to potentiate tissue plasminogen activator-induced reperfusion. However, there is growing evidence that thrombin may also have extravascular effects that influence ischemic brain injury. Male Sprague-Dawley rats were subjected to either 90 minutes of temporary middle cerebral artery (MCA) occlusion or sham operation to examine thrombin and protease activated receptor-1 (PAR-1) expression. In another set of rats, the MCA was occluded for 90 minutes and 10 U of hirudin or the same volume of vehicle was injected into the caudate followed by reperfusion for up to 28 days, to test the effects of local thrombin inhibition on ischemic damage, neurologic outcome and cerebral blood flow (CBF). Thrombin immunoreactivity was increased in the ischemic caudate at 4 and 24 hours, whereas PAR-1 expression was unchanged. Hirudin reduced infarct volume in the caudate at 24 hours (79 ± 41 vs. 115 ± 20 mm3, P < 0.05) and resulted in a larger residual tissue volume in the caudate at 28 days (17.6 ± 3.9 vs. 11.8 ± 6.3 mm3, P < 0.05). Hirudin treatment also had a beneficial effect on body weight and ameliorated neurologic deficits tested by forelimb placing and forelimb use asymmetry during 28 days survival. These beneficial effects of hirudin were not associated with improved regional CBF during reperfusion. These results suggest that, in addition to their effects on coagulation and circulation, thrombin inhibitors also have direct neuroprotective properties and may be considered in stroke therapy.
The serine protease thrombin has important roles in vascular events including hemostasis, inflammation, and angiogenesis. The vascular effects of thrombin have led to considerable work on the use of thrombin inhibitors to prevent the occurrence of cerebral ischemia and to potentiate reperfusion after an ischemic event (Hara et al., 1994; Kawai et al., 1996; Mikulski et al., 2001; Mima et al., 2000; Morris et al., 2001; Ohyama et al., 2001). However, it is becoming clear that thrombin is also involved in many extravascular events, including neuronal development, function, and degeneration. Such cellular actions of thrombin have been attributed to activation of some members of the protease-activated receptor (PAR) family. Both neuronal and glial cells express PAR transcripts as well as the thrombin precursor prothrombin (Dihanich et al., 1991; Niclou et al., 1994; Striggow et al., 2001; Wang et al., 2002). Interestingly, PARs are found in brain regions that are particularly vulnerable to ischemia (Striggow et al., 2001; Weinstein et al., 1995).
Disruption of the blood–brain barrier after cerebral ischemia may result in the entry of prothrombin (and other clotting factors) into brain from blood. In addition, brain cells may also be a source of thrombin under ischemic conditions (Riek-Burchardt et al., 2002). Our laboratory has shown that thrombin activity and prothrombin-mRNA expression increases in the brain after focal cerebral ischemia (Xi et al., 2002). The effects of thrombin may depend on concentration and the physiologic state of the brain. Low concentrations of thrombin are neuroprotective against experimental ischemia (Striggow et al., 2000), but high concentrations exert cytotoxic effects (Donovan et al., 1997; Jiang et al., 2002; Striggow et al., 2000). Furthermore, moderate concentrations of thrombin that are normally nontoxic decrease neuronal survival under ischemic conditions (Striggow et al., 2000; Xi et al., 2002).
To further elucidate the potential extravascular effects of thrombin in stroke, we examined the expression of thrombin and the main thrombin receptor, PAR-1, in a rat model of transient focal cerebral ischemia. We then tested the effects of an intracerebral injection of hirudin (a direct thrombin inhibitor) on tissue damage, neurologic outcome, and regional CBF.
MATERIALS AND METHODS
Animal model and experiment design
The University of Michigan Committee on the Use and Care of Animals approved the protocols for the animal studies. A total of 64 male Sprague-Dawley rats (275 to 325 g; Charles River Laboratories, Portage, MI, U.S.A.) were used. The animals were fasted for 4 hours before surgery but had free access to water. Anesthesia was induced by inhalation of 5% isoflurane in a 70% nitrous oxide, 30% oxygen mixture and maintained by 1.5% isoflurane administered through a facemask. The tail artery was cannulated percutaneously for monitoring pressure and blood sampling to measure PaO2, PaCO2, pH, hematocrit, and glucose. Rectal temperature was maintained at 37.5°C using a feedback-controlled heating pad. Transient focal cerebral ischemia was induced using the intraluminal suture method (Memezawa et al., 1992). In brief, a 23-mm segment of 3-0 nylon monofilament suture with the tip rounded by flame was inserted into the stump of the left common carotid artery and advanced into the internal carotid artery approximately 19 or 20 mm from the bifurcation to occlude the ostium of the middle cerebral artery (MCA). The suture was removed after 90 minutes and the animal was allowed to recover. The sham operation was identical except that the intraluminal suture was not inserted.
To examine thrombin and PAR-1 expression, 22 rats underwent transient MCA occlusion or sham operation and were killed 4 or 24 hours (n = 11 at each time point) after ischemia onset (2.5 and 22.5 hours of reperfusion). To test the effects of hirudin, 42 animals were used. After the occlusion of MCA, rats were positioned in a stereotactic frame (Kopf Instruments, Tujunga, CA, U.S.A.) under isoflurane anesthesia. Starting 15 minutes before reperfusion, 10 U of hirudin (Sigma, St. Louis, MO, U.S.A.) or an equal volume (2 × 2.5 μL) of saline was injected into the left caudate at a rate of 1 μL/min through a 26-gauge blunt needle using a microinfusion pump (World Precision Instruments, Sarasota, FL, U.S.A.). Two injections were given. The coordinates for the injection sites were 0.2 mm anterior, 3.5 mm lateral, 4 mm (2.5 μL) and 5 mm (2.5 μL) ventral to the bregma. Sixteen of these animals (hirudin n = 8, vehicle n = 8) were killed 24 hours after ischemia onset to measure infarct volume. Another 16 (hirudin n = 8, vehicle n = 8) were used for behavioral testing and body-weight measurement at days 1, 3, 7, 14, 21, and 28 and then killed for histologic examination. In the remaining 10 animals (hirudin n = 5, vehicle n = 5), CBF was measured at 4 hours after ischemia onset (2.5 hours of reperfusion).
Immunohistochemistry
Animals (n = 10, two or three in each group) were perfused with saline followed by 4% paraformaldehyde. The brains were removed and immersed into 4% paraformaldehyde for 6 hours followed by 25% sucrose for 3 days at 4°C. The tissue was frozen, embedded in O.C.T. compound (Sakura Finetek, Torrance, CA, U.S.A.) and 20-μm sections were taken at 1,000-μm intervals on a cryostat. Sections were stained for thrombin using the avidin-biotin complex technique (Xi et al., 1999). For PAR-1, a rhodamine-conjugated secondary antibody was used and the sections were examined under a fluorescence microscope (Xi et al., 1999). The following primary antibodies were used: polyclonal antithrombin antibody (1:400; Affinity Biologicals, Hamilton, Ontario, Canada), polyclonal anti–PAR-1 antibody (1:400; Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.). Normal sheep and rabbit immunoglobulin G were used as negative controls (Vector Laboratories, Burlingame, CA, U.S.A.).
Western blot analysis
Animals (n = 12, three in each group) were perfused with saline. The brain stem, hippocampus, thalamus, and the portion inferior to rhinal fissure were removed. Four samples (ipsilateral or contralateral to the MCA occlusion) were taken from the remaining tissue using 5-mm and 7-mm cork borers (Martz et al., 1990). The caudate and core cortex samples were taken from the striatum and cortex underlying the MCA immediately distal to the region of occlusion. The intermediate region was a ring of tissue surrounding the core, whereas the outer region consisted of the remainder of the cortical tissue. Western blot analysis was performed as previously described (Xi et al., 1999). Briefly, 50 μg protein from each sample was separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to a Hybond-C pure nitrocellulose membrane (Amersham Biosciences, Piscataway, NJ, U.S.A.). Rat plasma (prothrombin), rat thrombin and human thrombin (Sigma) were used as positive control. Membranes were probed with the same primary antibodies used for immunohistochemistry (1:1,000 antithrombin antibody, 1:1,000 anti–PAR-1 antibody) followed by peroxidase-conjugated antisheep and antirabbit secondary antibodies (1:2,500, Jackson ImmunoResearch Laboratories, West Grove, PA, U.S.A.). Antithrombin antibody that had been incubated with human thrombin was used as negative control. The antigen-antibody complexes were visualized with a chemiluminescence system (Amersham Biosciences) and exposed to Kodak X-OMAT film. The relative optical densities of bands were analyzed with the NIH Image 1.62 software (National Institute of Health, Bethesda, MD, U.S.A.).
Physiologic variables
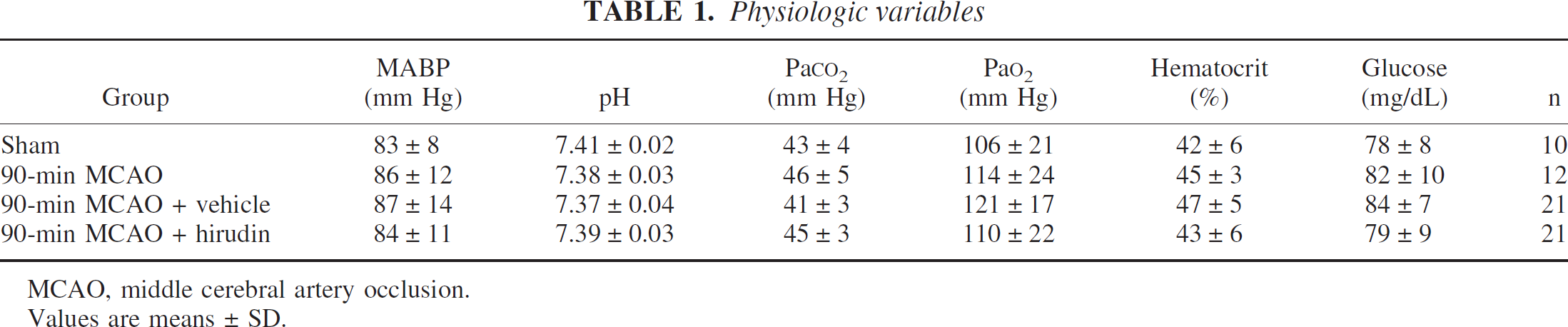
MCAO, middle cerebral artery occlusion.
Values are means ± SD.
Morphometric measurement of tissue damage
To identify the infarct area at 24 hours after ischemia onset (22.5 hours of reperfusion), fresh brain tissue was cut into 2-mm-thick coronal sections and incubated in 2% 2,3,5-triphenyltetrazolium chloride (TTC). To examine brain injury at 28 days after ischemia, the volume of residual tissue in the caudate and cortex was measured on histologic sections. These were prepared as described for immunohistochemistry and stained with hematoxylin and eosin. The area of infarction or remaining tissue was analyzed with the NIH Image 1.62 software, and the volume was calculated by multiplying the distance between sections.
Behavioral tests
The tester (Y. H.) was highly experienced and blind to the condition of the animals. Three behavioral tests were used: a forelimb placing test, a forelimb use asymmetry (cylinder) test, and a corner turn test. The animals were pretested 1 day before the surgical procedure to rule out any existing asymmetry and to obtain control values. The details and advantages of these tests have been reviewed elsewhere (Hua et al., 2002; Schallert et al., 2000).
Forelimb placing test. Animals were held by their torsos, allowing the forelimb to hang free. Independent testing of each forelimb was induced by brushing the respective vibrissae on the corner edge of a countertop. Intact animals place the forelimb quickly onto the countertop. Depending on the extent of injury, placing of the forelimb contralateral to the injury may be impaired. In the experiments, each rat was tested 10 times for each forelimb, and the percentage of trials in which the rat placed the right forelimb was used as the forelimb placing score.
Forelimb use asymmetry test. Forelimb use during exploratory activity was analyzed by videotaping rats in a transparent cylinder. The independent use of the left (L) or right (R) forelimb, or the simultaneous use of both (B) forelimbs for contacting the cylinder wall during a full rear or for alternating lateral stepping movements were recorded (a total of 20 per animal). A single overall forelimb use asymmetry score was calculated as follows: [(L – R) / 20] × 100.
Corner turn test. The rat was allowed to proceed into a corner, the angle of which was 30°. To exit the corner, the rat could turn either to the left or the right, and the turns involving full rearing along either wall were recorded (a total of eight per animal). Depending on the extent of injury, rats may show a tendency to turn to the side of the injury. The percentage of left turns was used as the corner turn score.
Cerebral blood flow measurement
A member of the team (S. R. E.) who was blind to the condition of the animals performed the CBF studies. Four hours after ischemia onset (2.5 hours of reperfusion), animals were reanesthetized and the regional CBF was determined by the indicator fractionation technique (Van Uitert and Levy, 1978) using [14C]-N-isopropyl-p-iodoamphetamine ([14C]IMP; American Radiolabeled Chemicals, St. Louis, MO, U.S.A.; (Williams et al., 1991). This method uses an intravenous bolus injection of a blood flow indicator followed by a constant rate of blood withdrawal through a femoral artery catheter to obtain the integral of the arterial isotope concentration. The withdrawal was started 5 seconds before injection of 5 μCi of [14C]IMP. Two minutes later, the animal was killed by decapitation and blood withdrawal was stopped. The sample of withdrawn arterial blood was bleached with 30% H202 before the addition of scintillation fluid and counting using a Beckman 3801 Liquid scintillation counter. A 3-mm-thick coronal brain slice was cut (4 mm from the frontal pole). The slice was divided into four samples: contralateral and ipsilateral caudate and contralateral and ipsilateral cortex. The samples were then digested in methylbenzethonium hydroxide before counting. The blood flow rates for the individual pieces of brain tissue were calculated using the following equation:
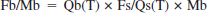
Where Fb is the brain blood flow, Mb is the brain mass (in grams), Qb(T) is the quantity of indicator in the tissue at the time T, Fs is the rate of blood withdrawal from t = 0 to t = T, and Qs(T) is the quantity of indicator present in the withdrawn blood at time T. The CBF is expressed as mL · 100 g−1 · min−1.
Statistical analysis
All quantitative data are presented as means ± SD. Volumes of tissue damage and Western blotting data were compared using the Mann-Whitney U test. Changes in body weight and behavioral testing data were compared using the Student's t-test. Differences are considered significant at the P < 0.05 level.
RESULTS
All values for MABP, blood pH, PaO2, PaCO2,, glucose, and hematocrit were in the normal physiologic range (Table 1).
There was increased thrombin immunostaining of parenchymal cells in the ischemic core compared with the contralateral hemisphere and sham-operated brains (Fig. 1A, B, and C). The negative control sections showed no staining (Fig. 1D). PAR-1 was detected in all brain regions by immunostaining and Western blotting but the expression was not altered by ischemia (Fig. 2A). Western blot analysis using antithrombin antibody showed a thrombin band at 37 kd and another band around 70 kd that may correspond to prothrombin or thrombin-antithrombin complexes. The positive controls showed high specificity of the antithrombin antibody for rat thrombin and prothrombin. The negative control membranes probed by antithrombin antibody that had been incubated with human thrombin showed no bands (Fig. 2B). Compared with the shams, thrombin in the ischemic caudate increased significantly at 4 hours (2.5 hours of reperfusion, 9,597 ± 1,888 pixels vs. 4,630 ± 626 pixels, P < 0.05, Fig. 2C) and 24 hours (22.5 hours of reperfusion, 10,391 ± 1,196 pixels vs. 5,733 ± 1,133 pixels, P < 0.05, Fig. 2D) after ischemia onset. Thrombin in the core cortex was increased at 24 hours (7,127 ± 3,270 pixels vs. 2,652 ± 437 pixels, P < 0.05) but was not altered at 4 hours (P > 0.05). The 70-kd band showed parallel increases to the thrombin band. Thrombin was detected in the intermediate, outer and contralateral regions but the blots were not significantly different compared with the sham controls.
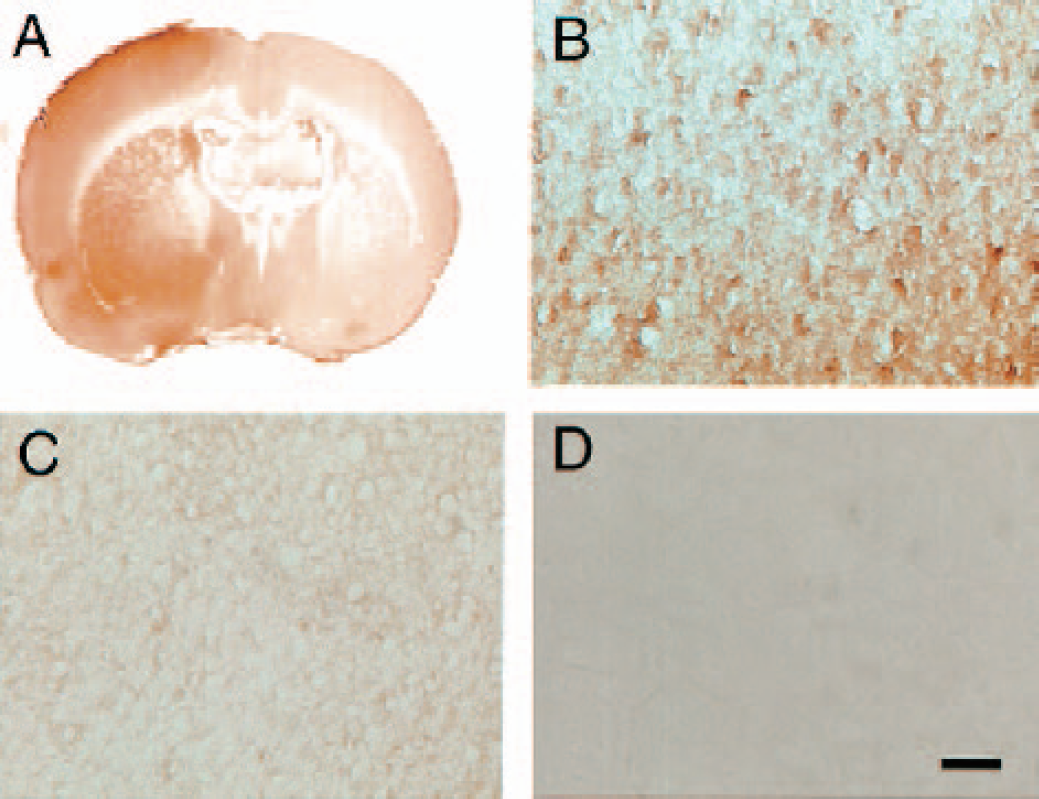
Thrombin immunoreactivity at 24 hours (90 minutes of MCA occlusion and 22.5 hours of reperfusion) after ischemia onset (
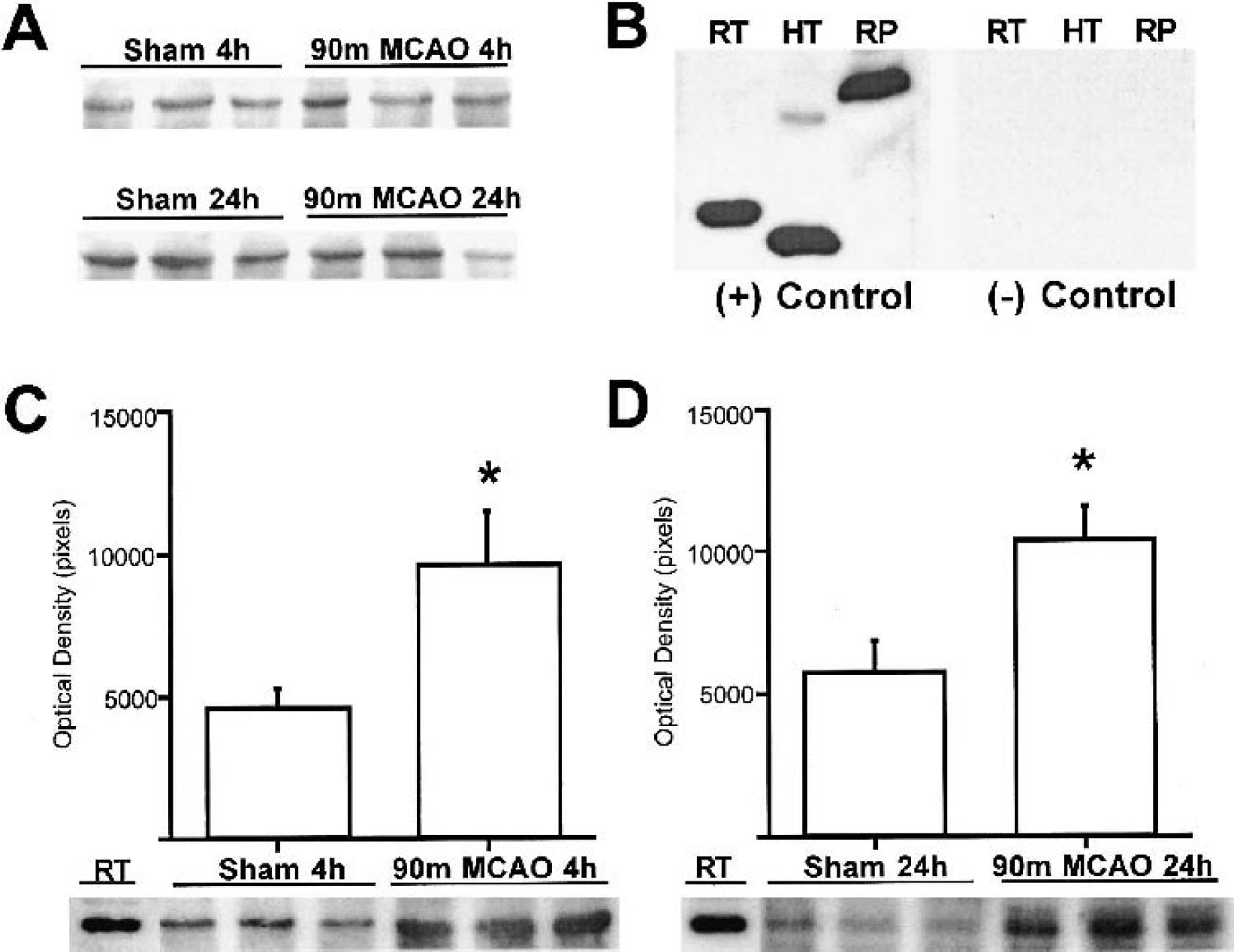
Western blot analysis of the ischemic caudate for thrombin and PAR-1 at 4 hours (90 minutes of MCA occlusion and 2.5 hours of reperfusion) and 24 hours (90 minutes of MCA occlusion and 22.5 hours of reperfusion) after ischemia onset. The PAR-1 expression was not altered by ischemia at 4 and 24 hours (
The intracaudate injection of hirudin decreased the infarct volume in the caudate detected by TTC staining at 24 hours after ischemia onset (79 ± 41 mm3 vs. 115 ± 20 mm3, P < 0.05; Fig. 3A). The cortical infarct volume was not altered by intracaudate hirudin injection (141 ± 108 mm3 vs. 162 ± 118 mm3, P > 0.05). The analysis of the hematoxylin and eosin-stained histologic sections from 28 days survival animals also revealed a significantly larger residual tissue volume in the ischemic caudate of hirudin-treated animals compared with the vehicle group (17.6 ± 3.9 mm3 vs. 11.8 ± 6.3 mm3, P < 0.05; Fig. 3B). The cortical residual volume was not significantly different at 28 days (P = 0.73). No hemorrhagic complications were noted in either hirudin-treated or vehicle-treated rats. In the 28-days survival experiment, the hirudin-injected animals also had better scores in behavioral tests and weight gain compared with the vehicle-injected animals. One animal in the vehicle treated group died at day 10 with severe neurologic deficits and progressive weight loss. The change in body weight was significantly better in the hirudin-treated group compared to vehicle-treated rats at days 7 (93% ± 7% vs. 81% ± 11% of initial, P < 0.05) and 28 (132% ± 17% vs. 106% ± 28% of initial, P < 0.05; Fig. 4A). Among the behavioral tests, the most significant improvement was in the forelimb placing score. By day 7, the placing score in hirudin-treated animals was 90 ± 10%, similar to pretest scores (100%), while the score for vehicle-treated rats was significantly less (5% ± 14%; P < 0.05). By day 28, the vehicle-treated animals were still more impaired (48% ± 51%) than hirudin-treated rats (100% ± 0%, P < 0.05; Fig. 4B). The hirudin-treated rats also scored better on the forelimb use asymmetry test (Fig. 4C). On the corner turn test, there was a tendency at all time points for the hirudin-treated animals to have a smaller deficit, but this did not reach significance (Fig. 4D).
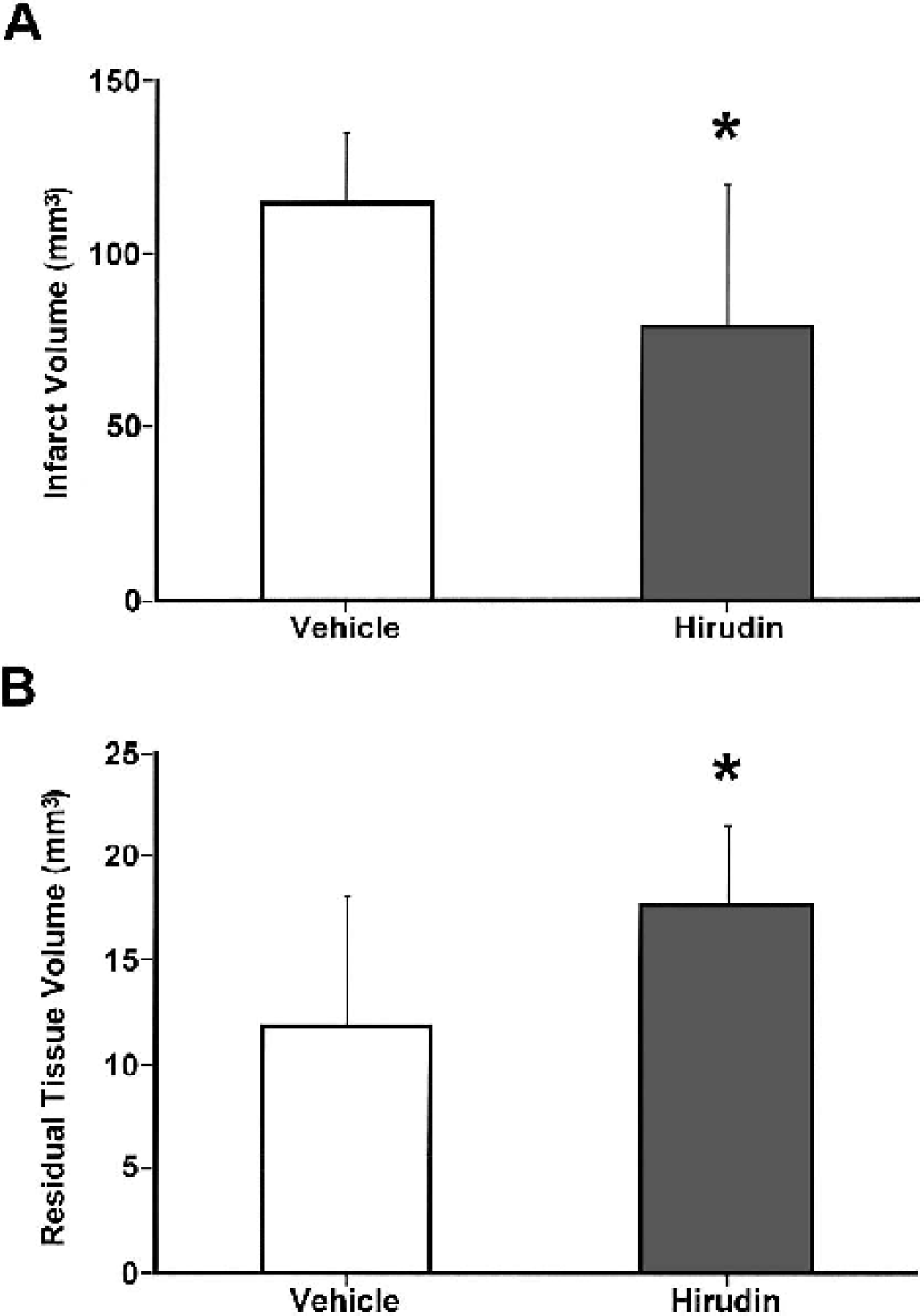
Attenuation of ischemic damage by hirudin. Intracaudate hirudin injection decreased infarct volume in the caudate detected by TTC staining at 24 hours (90 minutes of MCA occlusion and 22.5 hours of reperfusion) after ischemia onset compared with the vehicle-injected animals (
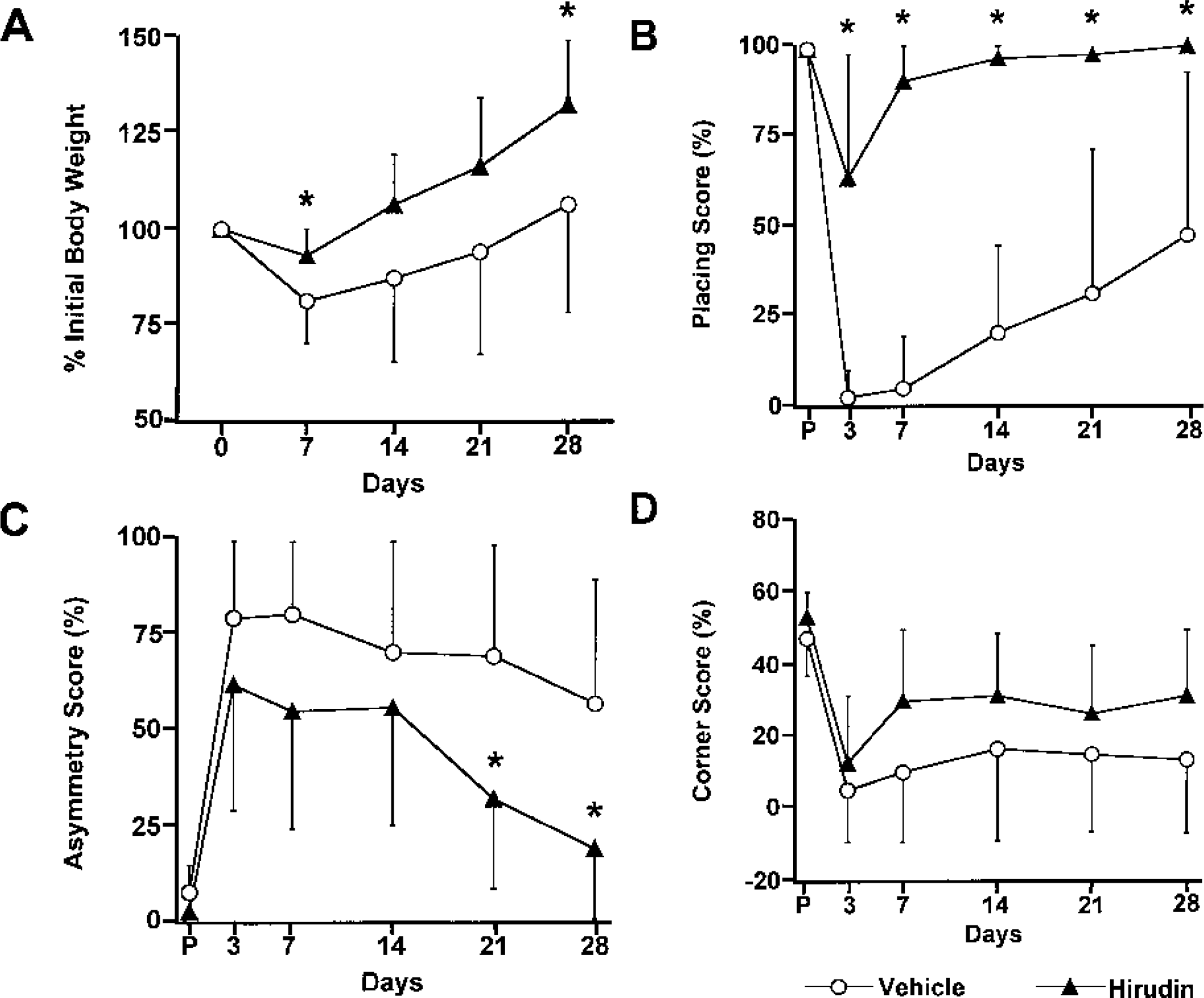
Body-weight change and behavioral test scores in the 28 days after 90-minute transient MCA occlusion. The animals injected with hirudin had a significantly better body weight at days 7 and 28 (
At 4 hours after ischemia onset (2.5 hours of reperfusion), the regional CBF decreased significantly in the ischemic caudate and cortex in both animal groups. The degree of hypoperfusion was not significantly different between the hirudin-treated and vehicle-treated animals (Fig. 5).
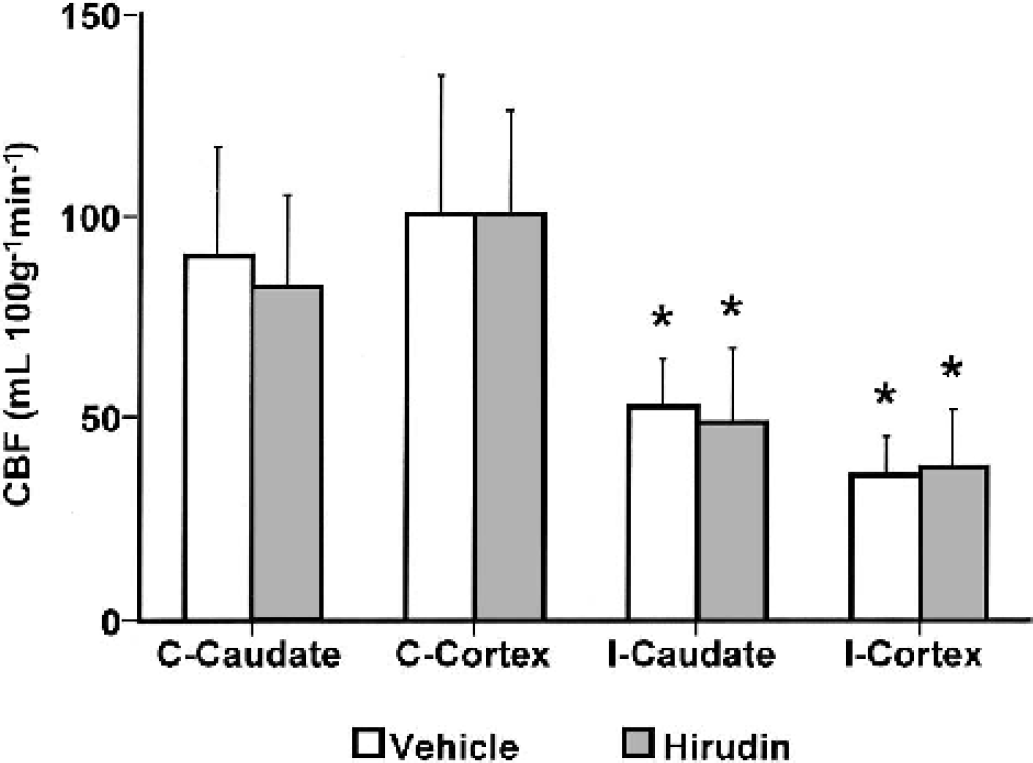
The regional CBF in caudate and cortex contralateral and ipsilateral to the site of MCA occlusion at 4 hours (90 minutes of MCA occlusion and 2.5 hours of reperfusion) after ischemia onset. Rats received an intracerebral injection of either hirudin or vehicle 15 minutes before reperfusion. There were no significant differences in regional CBF between hirudin-treated and vehicle-treated groups, with both groups showing hypoperfusion in the ipsilateral caudate and cortex compared to contralateral hemisphere. C, contralateral; I, ipsilateral. Values are means ± SD; *P < 0.05 vs. contralateral hemisphere.
DISCUSSION
The present study shows that thrombin expression increases after transient focal cerebral ischemia and that intracerebral injection of the thrombin inhibitor hirudin attenuates tissue damage and neurologic deficits after transient focal cerebral ischemia. The site of injection and the fact that there was no apparent alteration in local CBF suggest that the hirudin-induced protection is extravascular.
Thrombin and PAR-1 after cerebral ischemia
The current study used Western blots and immunohistochemistry to illustrate an upregulation of thrombin in the brain parenchyma after transient focal cerebral ischemia in the rat. Western blots were used to examine antibody specificity and to quantify thrombin upregulation. The antibody that was used recognizes both thrombin and prothrombin, and this is the probable explanation of the two bands detected in the ischemic brain: a thrombin band at 37 kd and a prothrombin band around 70 kd. However, it is possible that the latter may correspond to complexes of thrombin with thrombin inhibitors (such as protease nexin 1). Both bands showed similar changes during ischemia and preabsorption of the antibody with thrombin blocked both high- and low-molecular-weight bands.
Immunohistochemistry was used to examine the spatial distribution of thrombin (prothrombin). These results showed a widespread upregulation of thrombin throughout the ischemic territory. They also showed that there was an upregulation of extravascular thrombin; i.e. the perfusion of the brain with saline was indeed sufficient to remove intravascular thrombin/prothrombin.
It is important to consider the source of thrombin production. Thrombin can be produced in the brain after ischemia with hemorrhagic transformation, or it may result from an influx of prothrombin from blood after blood–brain barrier breakdown following cerebral ischemia. The concentration of prothrombin in plasma is about 1 to 5 μmol/L, suggesting that thrombin concentrations could reach very high levels in the brain parenchyma after cerebral ischemia. However, thrombin may also increase in the brain because of parenchymal production. Our laboratory has shown that prothrombin-mRNA and thrombin activity also increase after focal cerebral ischemia (Xi et al., 2002).
There is growing evidence that thrombin receptor activation is involved in brain pathology (Donovan et al., 1997; Smirnova et al., 1998; Striggow et al., 2000; Xi et al., 2003). Although the primary role of thrombin in hemostasis is through cleaving fibrinogen to fibrin and inducing platelet aggregation, other important cellular activities of thrombin may be related to thrombin receptor activation. Three protease-activated receptors PAR-1, PAR-3, and PAR-4 have been identified as thrombin receptors (Coughlin, 2000). Thus, thrombin signaling can be regulated by changes in the intracerebral thrombin concentration and/or by alterations in the expression of PAR subtypes. The current study shows that thrombin immunoreactivity increases in the ischemic core with constant expression of PAR-1 after transient focal cerebral ischemia. Similarly, Riek-Burchardt et al. (2002) found that global cerebral ischemia increases prothrombin mRNA in the adult rat hippocampus without altering the expression of PAR-1 mRNA. However, oxygen-glucose deprivation has been reported to increase PAR-1 mRNA expression in hippocampal slice cultures from juvenile rats (Striggow et al., 2001), suggesting that alterations in PAR-1 expression could be involved in some forms of ischemic brain injury. It is also possible that PAR-3 and PAR-4 levels may be changed by transient MCA occlusion.
Thrombin as a therapeutic target
Direct antithrombin drugs proved to be effective in preventing rethrombosis of the cardiac vessels and are becoming popular in cardiovascular units. Several researchers have examined the efficacy of antithrombin treatment in cerebral ischemia. Systemic thrombin inhibition attenuated neurodegeneration and brain edema formation after transient forebrain ischemia (Mima et al., 2000; Ohyama et al., 2001). Direct thrombin inhibitors decreased the number of microthrombi and the infarct volume, and improved neurologic outcome in thrombotic and embolic models of focal cerebral ischemia (Hara et al., 1994; Kawai et al., 1996; Mikulski et al., 2001; Morris et al., 2001). The effects of direct thrombin inhibitors in nonthromboembolic models of focal cerebral ischemia have not previously been tested. The focus of these previous experiments was the potential vascular effects of thrombin inhibition in cerebral ischemia. In all the experiments, intravascular thrombin inhibitors reversed the decrease in CBF and the neuroprotection was attributed to the improvement of microcirculation through inhibition of fibrin formation, platelet aggregation, and vasoconstriction. However, the improvement in the CBF and brain edema was not enough to explain the degree of neuroprotection in models of forebrain ischemia (Ohyama et al., 2001).
An alternate hypothesis, that extravascular thrombin might be involved in ischemic brain damage, has received little attention. Direct infusion of thrombin into the rat caudate nucleus induces brain edema, reactive gliosis, infiltration of inflammatory cells, proliferation of mesenchymal cells, and angiogenesis, resembling the events that occur after injury to the CNS (Lee et al., 1996; Nishino et al., 1993). It has been shown that thrombin can potentiate N-methyl-D-aspartate receptor activation (Gingrich et al., 2000). A thrombin concentration of 500 nmol/L is needed to exert degenerative effects in neuronal cultures. Although the current study shows an upregulation in thrombin after cerebral ischemia, it may be questioned whether such high concentrations are reached. However, even much lower levels of thrombin decrease neuronal survival after oxygen-glucose deprivation (Striggow et al., 2000). In addition, a previous study in our laboratory has shown that neither intracerebral injection of 1 U of thrombin nor 30 minutes of transient MCA occlusion increases brain water content, but the combination does result in significant brain edema (Xi et al., 2002). Therefore, it appears that under ischemic conditions thrombin does not need to reach high concentrations to unleash its deleterious effects.
The current study used intracerebral injection of hirudin to specifically address the question of whether extravascular effects of thrombin might influence ischemic brain injury. Hirudin, a polypeptide produced by the leech Hirudo medicinalis, inactivates thrombin by reversible tight complex formation (Markwardt, 2002). It has a molecular weight of 10 kd and does not cross the intact blood–brain barrier. It has been shown that intracerebroventricular infusion of hirudin increased the survival of hippocampal neurons after global cerebral ischemia in gerbils (Striggow et al., 2000). In this study, we used a hirudin dose of 10 U injected into the ischemic caudate just before reperfusion (1 U of hirudin neutralizes 1 U of thrombin). The dose of hirudin was based on the data showing that a thrombin concentration of 10 U/mL decreases the neuronal survival under hypoxic conditions (Striggow et al., 2000). Hirudin reduced the size of the caudate infarct identified by TTC staining 24 hours after MCA occlusion. Early histologic data have limitations in determining neuroprotection in animal models of stroke (Corbett and Nurse, 1998). Because of this, a further set of experiments was undertaken to examine long-term behavioral deficits, body weight changes, and histology. Thrombin inhibition with intracerebral injection of hirudin improved all three of these parameters: behavioral deficits, body weight, and caudate tissue loss. The behavioral tests used are not altered by repeated testing, do not require training, aversive motivation, or food deprivation, and appear to be very useful in assessing sensorimotor outcome in unilateral rat models of stroke (Hua et al., 2002; Schallert et al., 2000). Ninety minutes of MCA occlusion and reperfusion results in partial damage to the lateral aspects of the sensorimotor cortex and far lateral caudate. In the vehicle-treated rat, the behavioral tests showed moderate to severe deficits acutely, and substantial, though incomplete, recovery by the end of 28 days.
Although the molecular weight of hirudin is such that it will not cross the intact blood–brain barrier, there is the possibility that some may pass from brain to blood if the blood–brain barrier is disrupted, as in focal cerebral ischemia. Experiments measuring CBF were performed to directly address the issue of whether any such leak might be sufficient to influence blood flow. There were no differences in regional CBF between hirudin-treated and vehicle-treated rats, which strongly suggests that the protective effects of hirudin were extravascular.
In conclusion, our findings support that in addition to effects on vascular biology, thrombin inhibition has direct neuroprotective effects on brain cells and can be considered in the treatment and prevention of thromboembolic and nonthromboembolic stroke. Further investigation to identify the second-messenger systems and the molecular signaling pathways activated by PARs in neurons and glia will be important to elucidate mechanisms of direct neuroprotection by thrombin inhibitors.