
Abstract
The levels of protein kinase C-γ (PKC-γ) and the calcium/calmodulin-dependent kinase II-α (CaMKII-α) were measured in crude synaptosomal (P2), particulate (P3), and cytosolic (S3) fractions of the neocortex of rats exposed to 1-hour and 2-hour middle cerebral artery occlusion (MCAO) and 2-hour MCAO followed by 2-hour reperfusion. During MCAO, PKC levels increased in P2 and P3 in the most severe ischemic areas concomitantly with a decrease in S3. In the penumbra, PKCγ decreased in S3 without any significant increases in P2 and P3. Total PKC-γ also decreased in the penumbra but not in the ischemic core, suggesting that the protein is degraded by an energy-dependent mechanism, possibly by the 26S proteasome. The CaMKII-α levels increased in P2 but not P3 during ischemia and reperfusion in all ischemic regions, particularly in the ischemic core. Concomitantly, the levels in S3 decreased by 20% to 40% in the penumbra and by approximately 80% in the ischemic core. There were no changes in the total levels of CaMKII-α during MCAO. The authors conclude that during and after ischemia, PKC and CaMKII-α are translocated to the cell membranes, particularly synaptic membranes, where they may modulate cellular function, such as neurotransmission, and also affect cell survival. Drugs preventing PKC and/or CaMKII-α translocation may prove beneficial against ischemic cell death.
During middle cerebral artery occlusion (MCAO) in the rat, blood flow to the perfused brain territory decreases and is dependent on blood supply through collateral vessels (Memezawa et al., 1992a). A gradual decrease of blood flow therefore occurs from peripheral tissue towards the ischemic core surrounding the occlusion site. The blood flow in the periphery, also called the penumbra (Astrup et al., 1981; Hakim, 1987), is sufficient to uphold tissue viability, and some regions will recover after reperfusion (Memezawa et al., 1992b). More severe ischemic regions will eventually succumb and after 24 to 48 hours, tissue infarction is evident (Memezawa et al., 1992a,b). The variability of blood flow along the periphery to the core axis, will eventually lead to variable oxygenation and subsequently to variable energy; i.e., adenosine triphosphate (ATP) production (Folbergrova et al., 1992). This in turn will have direct consequences on ion homeostasis, evident as transient depolarization of the cell membrane, more frequent and prolonged in the core than in the penumbra (Kristian et al., 1998). The difference between energy production and dissipation of ionic gradients, in particular the calcium ion gradient, will activate intracellular mechanisms among other types of cell signaling, which may be protective or detrimental.
During and after transient global ischemia, cell signaling is severely disturbed (Ohtsuki et al., 1996; Shamloo and Wieloch, 1999; Takagi et al., 1997). In the vulnerable CA1 region of the hippocampus, protein kinase C-γ (PKC-γ) and the calcium/calmodulin-dependent kinase II (CaMKII) are persistently translocated from the cytosol to the cell membranes (Wieloch et al., 1991), and there is also a persistent elevation of tyrosine-phosphorylated proteins (Hu and Wieloch, 1993, 1994). In focal ischemia, it has been shown that both PKC (Crumrine et al., 1992) and CaMKII activity (Hanson et al., 1994; Waxham et al., 1996) are markedly changed, suggesting that cell signaling is aberrant in ischemic tissue.
The Ca2+ /phospholipid activated protein kinase C (PKC) is a central protein in neuronal metabolism, function, and survival (Dekker and Parker 1994; Nishizuka, 1986, 1992, 1995). PKC-γ requires phosphatidylserine, diacylglycerol, and Ca2+ for complete activation (Liu, 1996; Nishizuka, 1995). It can therefore be envisaged that PKC is differentially affected by variations in calcium levels as seen after MCAO. The importance of PKC for cell survival and plasticity is demonstrated in PKC-γ knockout mice, which develop normally but with a decreased capacity to induce long-term potentiation (Abeliovich et al., 1993). Also, staurosporine, a protein kinase inhibitor, reduces ischemic cell death in both rat and gerbil (Hara et al., 1990). In addition, substances that diminish PKC translocation to the cell membranes are neuroprotective (Karpiak et al., 1990). Intraischemic neuroprotective hypothermia (Boris Moller et al., 1989; Busto et al., 1987) also prevents the translocation of PKC (Busto et al., 1994; Cardell et al., 1991). The translocation of PKC-γ to membranes, which is also accompanied by a net and transient increase in its activity (Domanska-Janik, 1996), seems to cause pathological cell signaling and may contribute to ischemic cell death.
Likewise, CaMKII is a protein kinase found in all brain regions (Colbran, 1992; Hanson and Schulman, 1992) that has central functions in cellular metabolism and survival (Schulman, 1988; Schulman et al., 1992). CaMKII is mainly found in the postsynaptic densities (Strack et al., 1997) and therefore has an important role in signal transduction. It is a major component of the postsynaptic densities but is found in high amounts in the cytosol (Hu et al., 1995a,b). When activated by calcium ions, CaMKII becomes autophosphorylated at Thr 286 of CaMKII-α (Busto et al., 1994; Miller et al., 1988). The influx of Ca2+ into the postsynaptic cells promotes translocation of CaMKII from the cytosol to the membrane of postsynaptic densities (Strack et al., 1997). It has been suggested that the translocation of CaMKII to membranes after cerebral ischemia may enhance neuronal firing and calcium influx (Hu et al., 1995a; Matsumoto et al., 2002).
Because both PKC and CaMKII are markedly affected during and after transient global ischemia, we sought to study these enzymes in a model of MCAO. We therefore investigated the levels and subcellular distribution of PKC and CaMKII in different cortical areas during and after MCAO. The areas studied were chosen to include tissue with different degrees of reperfusion; i.e., both that of the penumbra and that of the ischemic core.
MATERIALS AND METHODS
Surgical procedures and experimental groups
This study consisted of the following experimental groups. For studies of subcellular fractionation: 1-hour MCAO (n = 3), 2-hour MCAO (n = 5), and 2-hour reperfusion after 2-hour MCAO (n = 6). For whole-tissue analysis: 2-hour MCAO (n = 6). The ethical committee at Lund University approved the transient MCAO procedures. The experiments were conducted as described previously (Memezawa et al., 1992a,b; Zhao et al., 1994). Male Wistar rats (Möllegaards Breeding Center, Copenhagen), weighing 310 to 350g, were housed with free access to water and fasted overnight before the experiments. The rats were anesthetized with an inhalation of 3% halothane in N2O:O2 (70%:30%), and were then intubated and ventilated on 1.0% to 1.5% halothane in N2O:O2 during surgery. Blood pressure, Pa
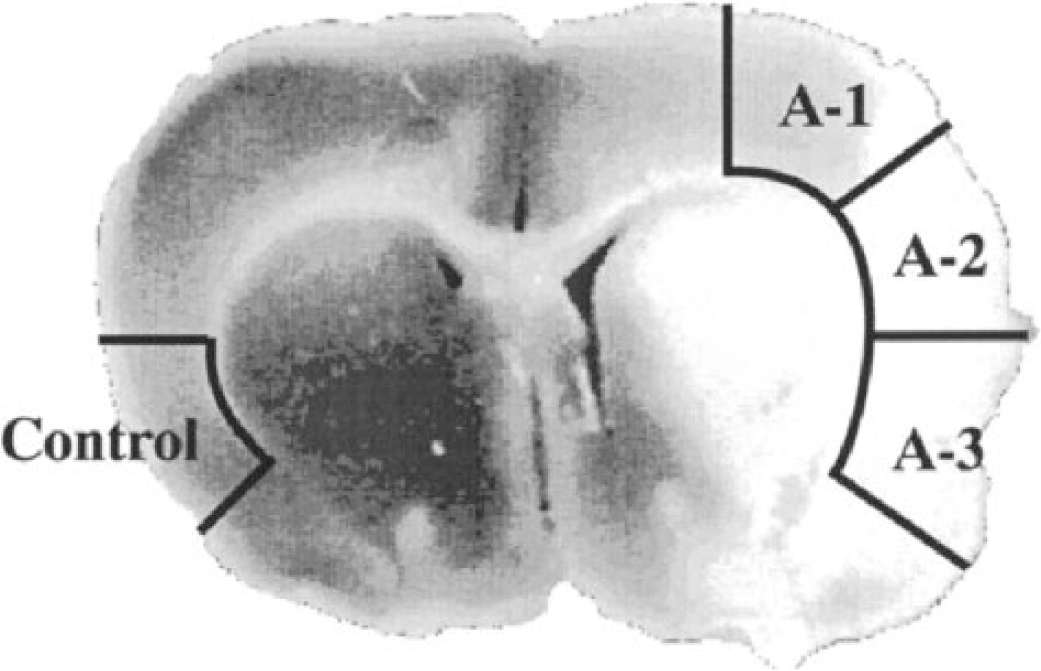
A coronal section of rat brain subjected to 2 hours of middle cerebral artery occlusion and 48 hours of reperfusion, stained with tetrazolium chloride to delineate the infarct area (white tissue). The areas where tissues were sampled are indicated. Control, contralateral to occluded hemisphere; A-1, mild ischemic area; A-2, more severely ischemic area; A-3, ischemic core.
Subcellular fractionation
Subcellular fractionation was performed as described earlier (Matsumoto et al., 2002). The tissue was sonicated twice for 10 seconds in 1:10 (wt/vol) homogenization buffer with the following composition: 50 mmol/L 3-[N-morpholino] propanesulfonic acid (pH 7.6), 2 mmol/L dithiothreitol, 0.1 mmol/L sodium orthovanadate, 3 mmol/L EGTA, 0.1 mmol/L PMSF, 20 μg/mL leupeptin, 5 μg/mL aprotinin, 10 μg/mL pepstatin, 0.5 mmol/L Mg(CH3COO)2, and 0.32 mol/L sucrose on ice. The homogenates were centrifuged at 800g for 10 minutes at 4°C followed by centrifugation of the supernatant at 9,200g for 15 minutes at 4°C. The resulting crude synaptosomal fraction (P2) was sonicated for 10 seconds in homogenization buffer containing 0.1% Triton X-100, and the supernatant was centrifuged at 165,000g for 1 hour at 2°C in a TL100.2 rotor. The supernatant fraction (S3), consisting of the cytosol, and the particulate fraction (P3) were reconstituted in homogenization buffer plus 1% Triton X-100. In a separate series of brains, tissue was homogenated as described previously but was not fractionated, and used for whole-tissue analysis. The protein concentration was determined (Lowry et al., 1951) before freezing the samples at −80°C for later analysis.
Electrophoresis and immunoblotting
Sodium dodecyl sulfate-polyacrylamide (8%) gel electrophoresis (SDS-PAGE) was used (Laemmli, 1970). The samples (12.5 μg protein for P2 and P3, 25 μg for S3) were mixed with a 5 × SDS sample buffer of 0.3-mol/L Tris/HCl (pH 6.8), 25% α- mercaptoethanol, 12% SDS, 25-mmol/L EDTA, 20% glycerol, and 0.1% bromphenol blue, and then boiled for 4 minutes. They were then subjected to SDS-PAGE at a constant current of 40 mA (stacking gel) and 45 mA (separating gel), after which the proteins were electrotransferred onto a polyvinylidene difluoride membrane (Bio-Rad transblot; pore size, 0.2 m) with constant current of 100 V/h (Towbin et al., 1979). After transfer, the polyvinylidene difluoride membrane was washed in Tris-buffered saline, containing 0.1% tween 20 (TBST), and then preincubated with a blocking solution of 3% bovine serum albumin in TBST for 1 hour at room temperature. The blot was incubated overnight with PKC-γ antibodies (anti-PKC-γ; Boehringer Mannheim, Germany), or CaMKII-α (anti-CaMKII-α; Boehringer Mannheim, Germany) in TBST containing 3% bovine serum albumin at 4°C, and then with a secondary antibody conjugated with horseradish peroxides in Tris-buffered saline containing 2% BSA for 1 hour at 4°C. The immunoreactivity of the antibody on the membranes was evaluated by the ECL+Plus Western blotting system (Amersham, UK), and the blots quantified with a DIANA-II CCD camera system and the TINA 2.0 program (Raytest Isotopenmessgeräte GmbH, Straubenhardt, Germany).
Statistical analysis
Data are presented as mean value with standard deviations (mean ± SD) or mean values with 95% or 99% confidence interval (95% or 99% CI; lower confidence limit to upper confidence limit). The changes in the levels of PKC and CaMKII were analyzed by comparing the 95% or 99% confidence intervals (P < 0.05 or P < 0.01) with the control value and analysis of variance followed by the post hoc Dunn-Bonferroni test (P < 0.05, P < 0.01). Physiologic data were analyzed with analysis of variance followed by Scheffé test (P < 0.05, P < 0.01).
RESULTS
Physiologic parameters
Physiologic parameters without for the whole-tissue analysis were same as our previous study (Matsumoto et al., 2002). Core temperature, blood pressure, arterial P
Physiologic parameters of rats subjected to middle cerebral artery occlusion
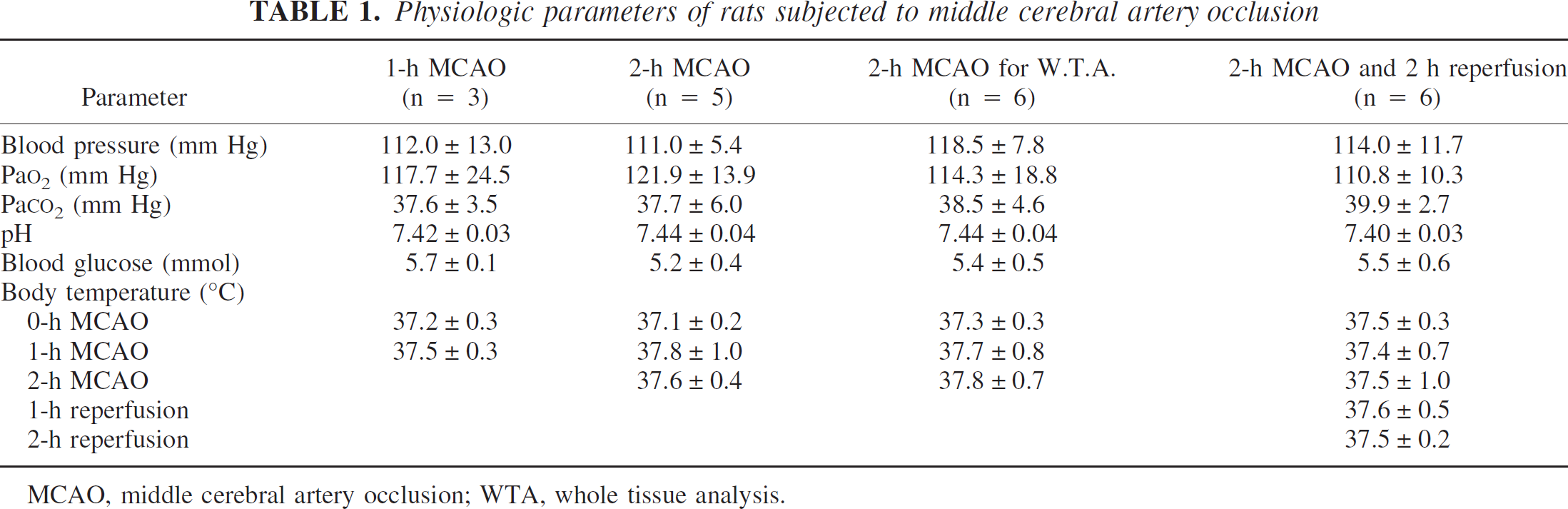
MCAO, middle cerebral artery occlusion; WTA, whole tissue analysis.
Changes in PKC levels
The tissue was sampled as indicated in Fig. 1, A-1 (weak ischemic penumbra), A-2 (strong ischemic penumbra), A-3 (ischemic core), and control (contralateral area of A-3) homogenized and fractionated into a synaptosomal (P2), particulate (P3), and cytosolic (S3) fraction. Figure 2 shows the levels of PKC at 1 and 2 hours of MCAO and 2 hours of reperfusion after 2-hour MCAO. The protein levels on the immunoblot were calculated and are presented as percentages of that in the control area.
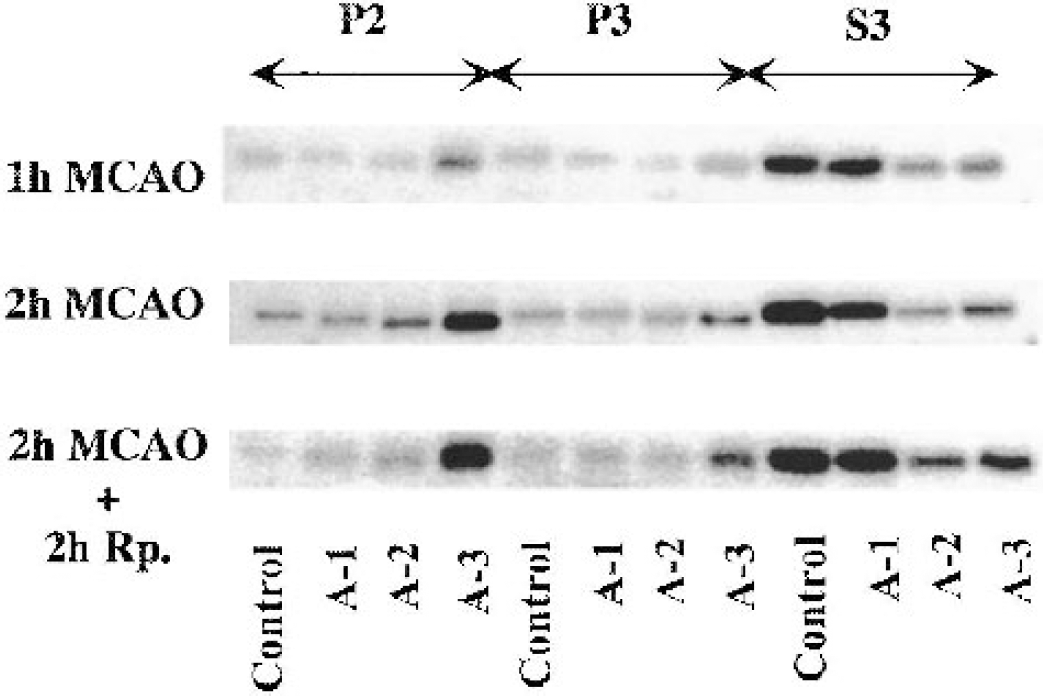
An immunoblot of PKC-γ in the crude synaptosomal fraction (P2), the particulate fraction (P3), and the cytosolic fraction (S3) from different brain areas as indicated in Fig. 1. The tissue was sampled at 1 and 2 hours of MCAO, and at 2 hours of reperfusion after 2 hours of MCAO.
At 1 hour of MCAO, there was a marked increase in PKC in the P2 fraction in A-3 and the level of PKC increased to 233% (99% CI: 115–351; P < 0.01). Similarly, PKC increased in P3, albeit to a lesser degree. In S3, the level of PKC decreased to 42% (99% CI: 4–80; P < 0.01) in A-2, and to 46% (99% CI: 0–94; P < 0.01) in A-3. There was no effect on the PKC levels in A-1 in any of the fractions at this time of ischemia (Fig. 3).
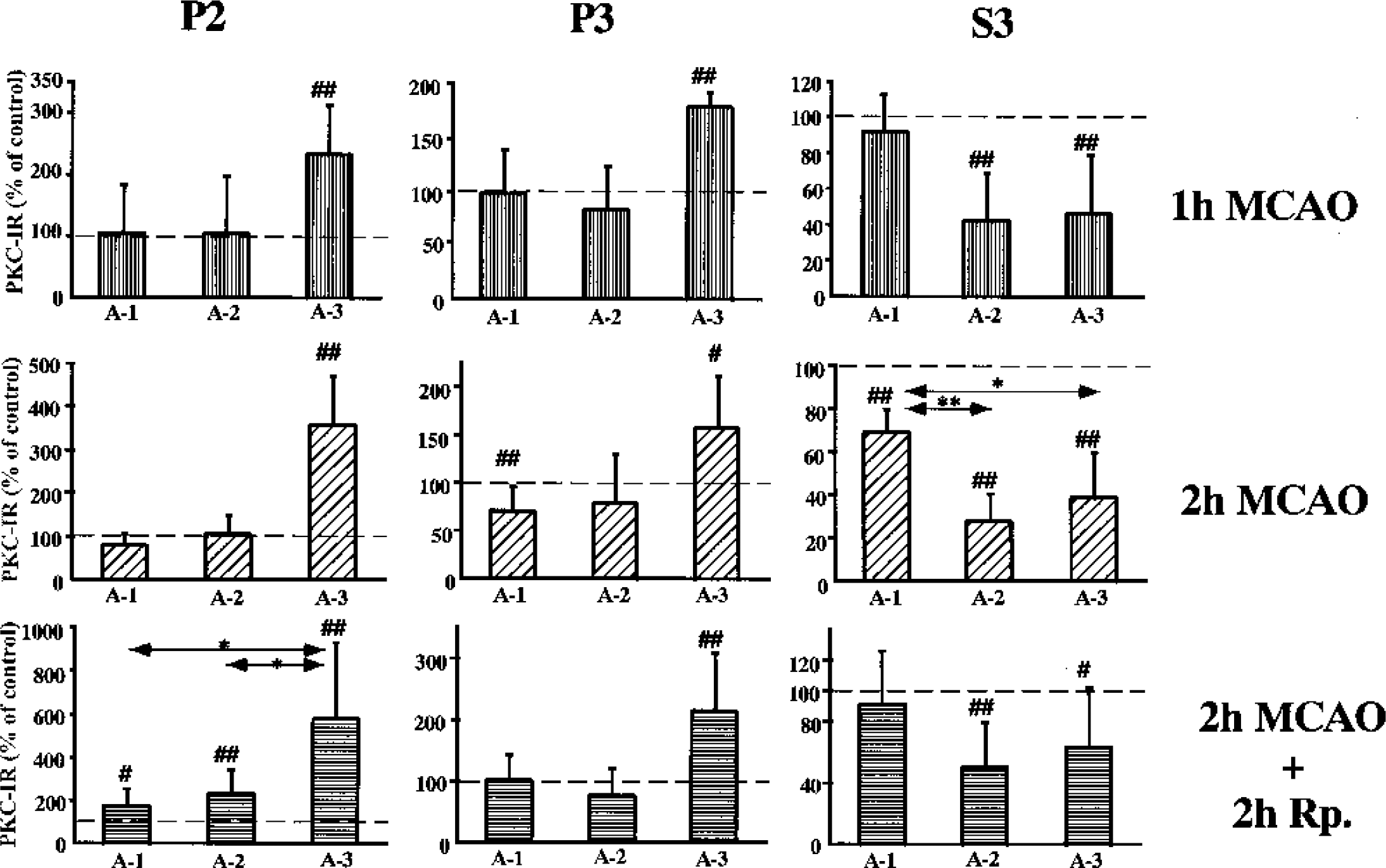
Changes in the levels of PKC-γ in the crude synaptosomal (P2), the particulate fraction (P3), and the cytosolic (S3) fraction from different brain regions as indicated in Fig. 1. The levels are presented as a percent of the control side. The values are means ± SD. Number signs denote statistical differences with the control side with 95% (#) and 99% (##) confidence intervals; *P < 0.05 and **P < 0.01, Dunn-Bonferroni test.
After 2 hours of MCAO, the level of PKC was further increased to 368% (99% CI: 249 to 483; P < 0.01) in P2 of A-3. However, a significant decrease to 70% (99% CI: 41–99; P < 0.01) was seen in P3 of A-1, whereas a significant increase to 158% (95% CI: 111–220; P < 0.05) was observed in A-3. In S3, the level decreased in all areas examined, albeit to different degrees, to 69% (99% CI: 56–82; P < 0.01), 28% (99% CI: 14–42; P < 0.01), and 39% (99% CI: 15–63; P < 0.01), in A-1, A-2, and A-3, respectively. The PKC levels were significantly less in A-2 (P < 0.01) and A-3 (P < 0.05) when compared with A-1 (Dunn-Bonferroni test) (Fig. 3).
At 2 hours of reperfusion after 2 hours of MCAO, the PKC levels significantly increased in P2 of all areas, particularly in A-3, to 581% (99% CI: 215–947; P < 0.01). In contrast, the level increased only to 214%, (99% CI: 118–312; P < 0.01) in P3 of A-3. In S3, the PKC level decreased to 51% in A-2 (99% CI: 22–80; P < 0.01) and to 64% in A-3 (95% CI: 33–95; P < 0.05) (Fig. 3).
The markedly decreased levels in S3 in both A-2 and A-3 suggest that PKC is depleted after 2 hours of MCAO. We therefore measured the total tissue level of PKC in another experimental series (Fig. 4). The level of PKC significantly decreased to 84% in the penumbra areas A-1 (99% CI: 74–94; P < 0.01) and to 68% in A-2 (99% CI: 51–85; P < 0.01), whereas there was no change in PKC levels in A-3. There were no differences in the total levels of PKC between the contralateral area and the A-1, A-2, or A-3 areas.
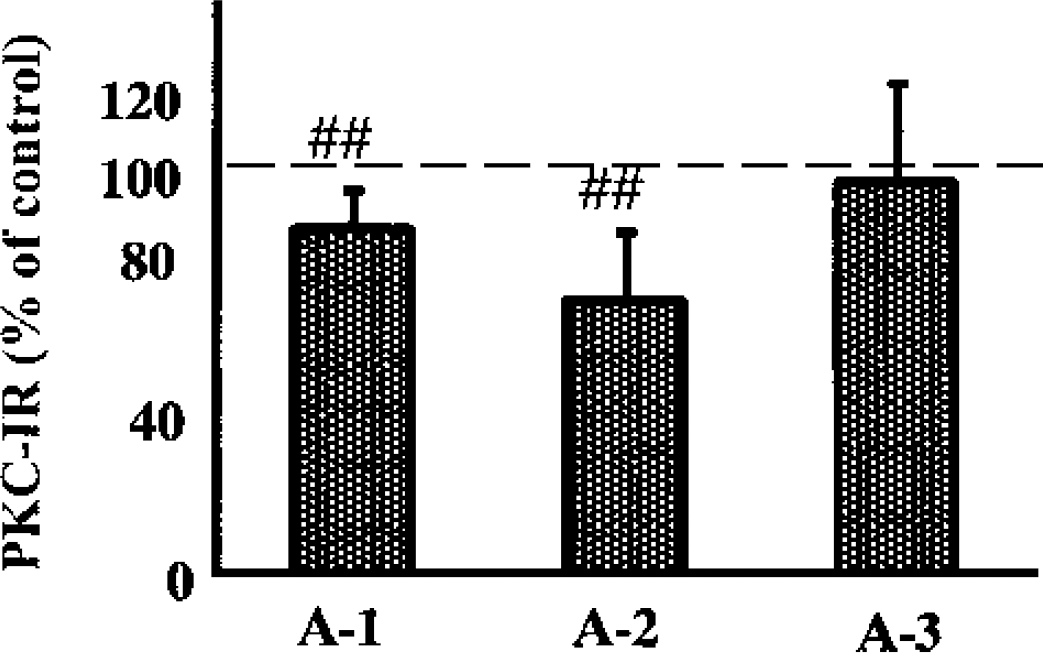
The levels of PKC-γ in whole-tissue homogenates from different brain regions after 2 hours of MCAO. The values are means ± SD. Number sign (##) denotes a statistical difference with the control side with 99% confidence intervals.
Changes in CaMKII levels
Figure 5 shows an immunoblot of CaMKII during 1-hour and 2 hour MCAO and 2-hour MCAO followed by 2-hour reperfusion. The level of CaMKII-α at 1-hour MCAO significantly increased in P2 of A-2 and A-3, whereas in P3 no significant difference was seen in any area. In S3, the level decreased in all the areas, especially in A-2 and A-3, where levels decreased to 20% (99% CI: 5–35; p<0.01) and 16% (99% CI: 3–29; p<0.01) of control levels, respectively (Fig. 6).
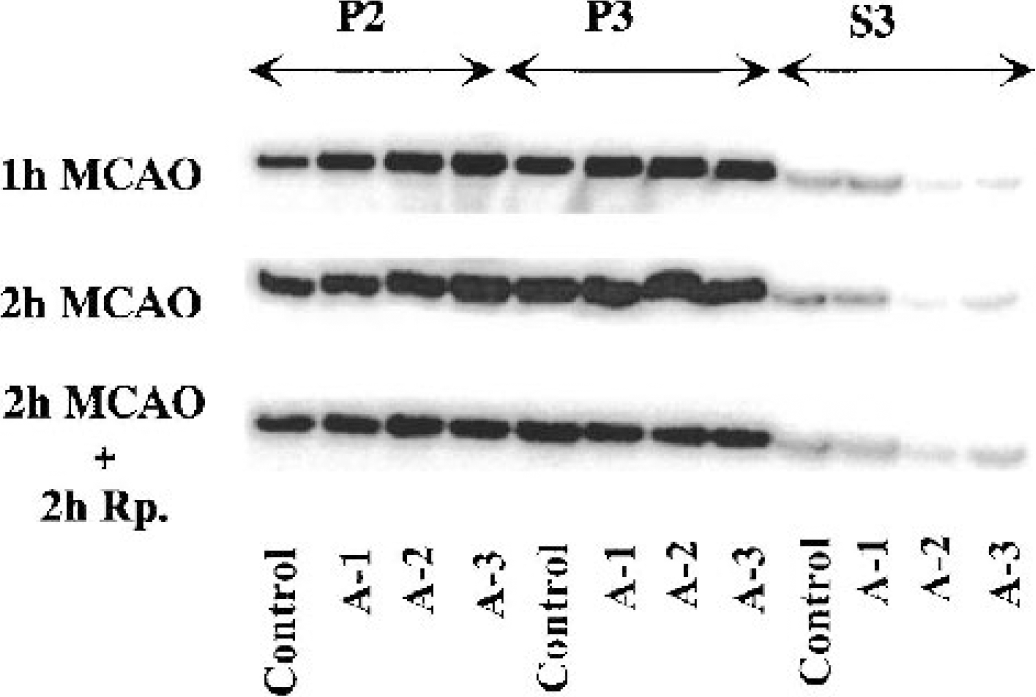
Immunoblots of CaMKII-α in the crude synaptosomal fraction (P2), the particulate fraction (P3), and the cytosolic fraction (S3) from different brain areas as indicated in Fig. 1. The tissue was sampled at 1 and 2 hours of MCAO, and at 2 hours of reperfusion after 2 hours of MCAO.
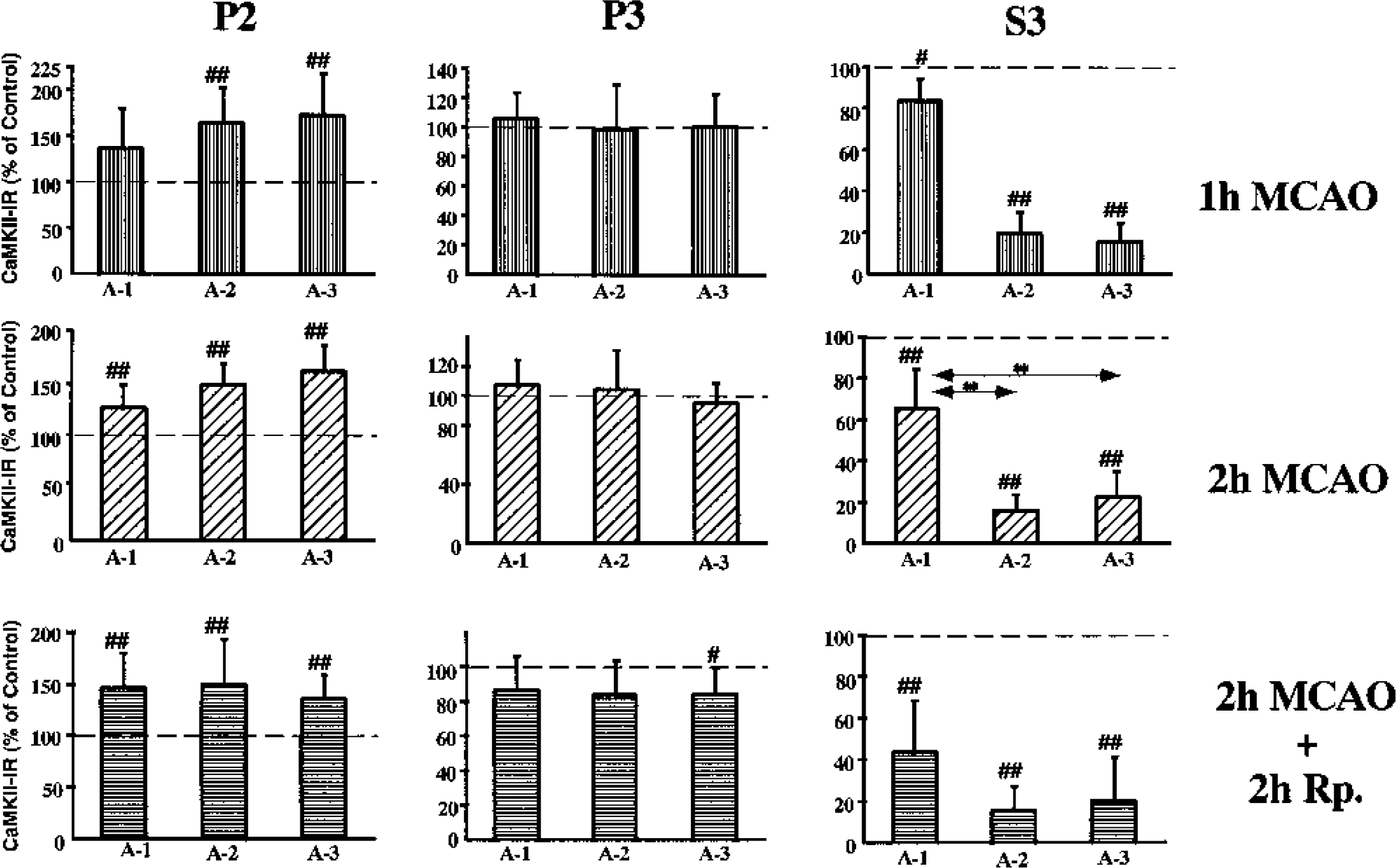
Changes in the levels of CaMKII-α from different brain regions as indicated in Fig. 1. Crude synaptosomal (P2) and particulate (P3) and cytosolic (S3) fractions from the different brain regions. The levels are presented as a percentage of the control side. The values are means ± SD. Number signs denote statistical differences with the control side with 95% (#) and 99% (##) confidence intervals, respectively; **P < 0.01, Dunn-Bonferroni test.
At 2 hours of MCAO, the CaMKII-α levels were significantly increased in P2 in all the ischemic areas, whereas no significant change in P3 was seen. In S3, the level decreased further, especially in A-2, where the level of CaMKII-α decreased to 17% (99% CI: 9–25; p<0.01), but also in A-3, where the level decreased to 23% (99% CI: 9–37; P < 0.01) (Fig. 6).
After 2 hours of MCAO at 2 hours of reperfusion, the CaMKII-α level in P2 increased in all areas by approximately 50%, whereas in P3 the level of CaMKII-α in A-3 was significantly decreased to 85% (95% CI: 73–97; P < 00.05) of the levels in the contralateral side. In S3, the levels simultaneously decreased to 44% (99% CI: 44–88; P < 00.01), 16% (99% CI: 9–25; P < 00.01), and 21% (99% CI: 9–37; P < 00.01) in A-1, A-2, and A-3, respectively (Fig. 6).
After ischemia and reperfusion, CaMKII-α levels in P2 increased in all the ischemic areas, and there was no significant difference between A-1, A-2, and A-3. In contrast, in P3 the CaMKII-α levels did not increase during MCAO, but rather a small decrease was observed at 2 hours of reperfusion. In S3, a persistent decrease was seen in all the ischemic areas, especially in A-2 and A-3. Unlike PKC, no significant change was observed in the total amount of CaMKII-α in whole-tissue homogenates (Fig. 7). There were no differences in the total levels of CaMKII between the control side and A-1, A-2, or A-3.
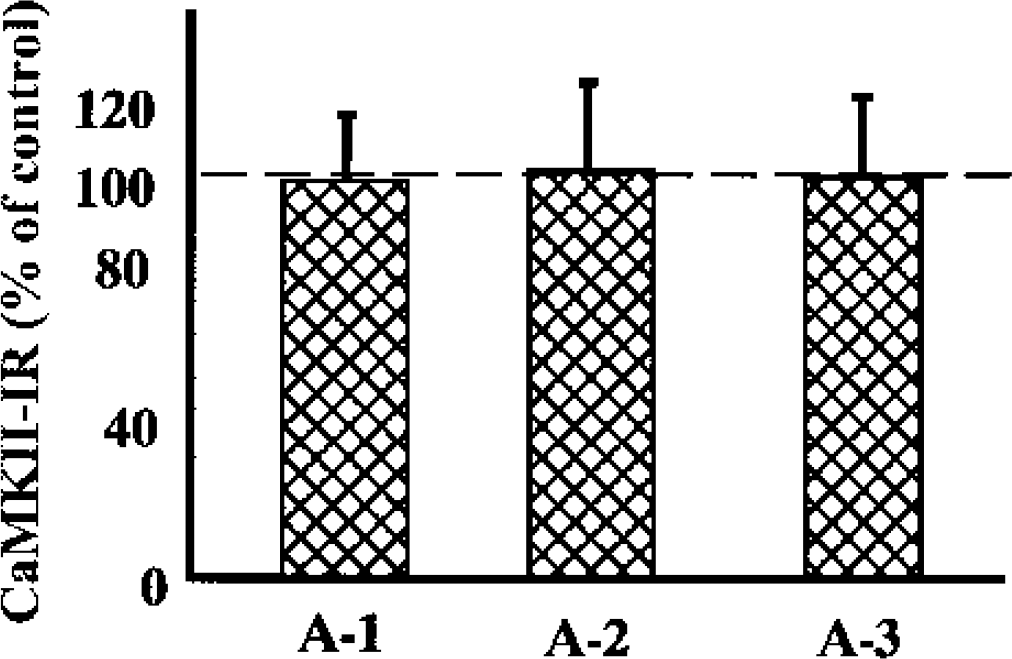
The levels of CaMKII-α in whole-tissue homogenates from different brain regions after 2 hours of MCAO.
DISCUSSION
In the ischemia model used in the present study, after 60 and 180 minutes of ischemia the cerebral blood flow decreased to approximately 40% of control levels in A-1, to 18% in A-2, and to 10% in A-3 (Memezawa et al., 1992a). We therefore can consider A-1 to be a mild ischemic area, A-2 a more severely ischemic area, and A-3 the ischemic core. The differences in tissue perfusion were also reflected in the differences in tissue ATP levels. In an area that is slightly larger than A-1, ATP levels decrease by approximately 35%, whereas in an area that is slightly larger than A-3, approximately 70% of ATP levels are lost (Folbergrova et al., 1992). Therefore, energy production in A-1 is still significant, which explains the transient recovery from ionic gradients seen when measured with ion-selective electrodes (Gido et al., 1997; Kristian et al., 1998). The variability in tissue ATP levels and calcium concentrations could also have contributed to the changes in PKC and CaMKII levels observed during and after MCAO in this investigation.
Effects of MCAO on CaMKII and PKC
Translocation of PKC, leading to firm binding of the protein to cell membranes, is stimulated by calcium ions (Nelsestuen and Bazzi, 1991). The marked translocation to the synaptosomal (P2) and particulate (P3) fraction in the ischemic core (A-3) and the subsequent decrease in the cytosol (S3) are probably due to the stimulating effect of the massive calcium influx occurring during ischemia (Silver and Erecinska, 1992). The translocation was not followed by a decrease in the total levels of PKC in the ischemic core (A-3) at the end of the ischemic episode (Fig. 4), indicating that the translocation is more likely a redistribution. In contrast, in the penumbra areas, A-1 and A-2, no marked increase in synaptosomal (P2) PKC levels was seen at 2 hours of MCAO, but a significant decrease was seen in both the particulate (P3) and cytosolic (S3) fractions. The total PKC levels also decreased in these fractions (Fig. 4), suggesting that PKC is degraded. Protein kinase C is a substrate of calcium-activated proteases, the calpains (Kishimoto et al., 1989; Saido et al., 1991). Because the calcium concentration is most certainly elevated in the ischemic core as well as in the penumbra area, it is less likely that calpains are responsible for decreases in PKC in the penumbra region. Apparently, the total levels decreased in areas where ATP production is maintained, suggesting that ATP-dependent proteolysis may occur. Because PKC is a substrate of the 26S proteasome (Favit et al., 1998), we propose that this protease complex is activated in the penumbra region and may contribute to cell death. Indeed the proteasome has been implicated in apoptosis (Naujokat and Hoffmann, 2002) and inhibition of the proteasome complex is neuroprotective (Phillips et al., 2000).
During reperfusion PKC is further translocated in the penumbra region, leading to an increase in PKC levels in the membranes while the levels decrease in the cytosol. The increase of PKC in the cell membranes of penumbra is in line with the notion that this area is recruited into the irreversible ischemic cell damage in a time-dependent manner. This might be recognized in the observations that 1,2 PDBU binding, an indicator of PKC levels, increases in these regions (Tohyama et al., 1997, 1998).
The changes in CaMKII are partly similar to those of PKC. However, there is no gross loss of protein in any of the brain regions (Fig. 7). CaMKII-α is translocated to cell membranes during MCAO. The translocation was observed in all ischemic regions but was most prominent in the ischemic core, A-3. During reperfusion the translocation persisted, and was further enhanced. Interestingly, the levels in the particulate fraction (P3), which encompasses the endoplasmic reticulum and plasma membrane, also decreased during reperfusion. Similarly, as for PKC, the data strongly suggest that the elevated calcium ion concentration in the ischemic areas stimulate CaMKII binding to synaptic membranes, suggesting that CaMKII is a more sensitive probe for ischemia than PKC.
The relation among PKC, CaMKII, and MCAO-induced cell death
We propose that CaMKII and PKC are translocated to cell membranes during MCAO, a processes stimulated by pathologically increased levels of calcium ions. The translocation may therefore contribute to the activation of detrimental processes leading to cell death. For example, hyperphosphorylation of the glutamate receptors by translocated PKC or CaMKII-α (Barria et al., 1997; Matsumoto et al., 2002; Soderling, 1996) may occur during reperfusion and with subsequent sensitization of the receptor to glutamate activation (Nellgard and Wieloch, 1992), increasing ion cycling across the membranes, including calcium ions. These changes in receptor sensitivity and calcium homeostasis could cause glutamate neurotransmission and calcium signaling to become detrimental during reperfusion.