
Abstract
Stroke patients with hyperglycemia (HG) develop higher volumes of brain edema emerging from disruption of blood–brain barrier (BBB). This study explored whether inductions of protein kinase C-β (PKC-β) and RhoA/Rho-kinase/myosin-regulatory light chain-2 (MLC2) pathway may account for HG-induced barrier damage using an in vitro model of human BBB comprising human brain microvascular endothelial cells (HBMEC) and astrocytes. Hyperglycemia (25 mmol/L
INTRODUCTION
Ischemic strokes develop through an interference with blood supply to the brain and continue to be the leading cause of morbidity in the world. Cerebral ischemia reperfusion triggers a series of events such as oxidative stress and inflammation, which collectively compromise the integrity and function of blood–brain barrier (BBB). 1 Disruption of the BBB in turn leads to severe neurologic deficits through aggravation of hemorrhagic transformation or vasogenic edema. 2 Although subtle increases in cerebral water content may be apparent within hours of stroke onset, the clinical signs of unattended brain edema peak typically during the following 1 to 5 days. Uncontrolled cerebral edema constitutes the leading cause of death within the first week after ischemic strokes.3, 4 As the incidence and severity of BBB damage is markedly higher in stroke patients with diabetes or acute hyperglycemia (HG) than those without, it is thought that HG has a pivotal role in the development and exacerbation of this defect.5, 6 However, the mechanisms involved remain largely unexplored and are of vital importance to develop efficacious novel therapeutic regimens.
The BBB is composed of brain microvascular endothelial cells, capillary basement membranes, and astrocyte end-feet. The restraining role of the BBB is in large part attributed to continuous presence of tight junctions, formed by several transmembrane and associated cytoplasmic proteins, notably occludin and zonula occludens-1 (ZO-1). 7 Given the pronounced roles of occludin and ZO-1 in tightening junctional complex and sustaining endothelial phenotype, 8 it is safe to suggest that any pathologic stimulus affecting the expression and localization of these proteins will profoundly affect the integrity of the BBB.
Hyperglycemia-mediated activation of protein kinase C (PKC), especially PKC-β, represents a key step in peripheral vasculopathies and indeed accounts for vascular leakage in organs that manifest ischemic injury- or diabetes-mediated damage such as heart and retina.9, 10 Naturally, attenuation of PKC-β activity has been coupled to amelioration of HG-induced retinal and glomerular microvascular complications to which inhibitions of ZO-1 and occludin translocation and the small GTPase RhoA post-translational modification appear to contribute.11, 12, 13 It is noteworthy in this context that the increased presence of PKC-β protein has also been documented in the brain infarcts of deceased ischemic stroke patients. 14
Guanosine triphosphate-binding proteins, in particular RhoA, have crucial roles in many cellular processes, including the regulation of endothelial barrier integrity and function.15, 16 Once activated, RhoA binds to its downstream effector Rho-kinase and as a consequence induce myosin-regulatory light chain-2 phosphorylation (p-MLC2) and actin stress fiber formation in a sequential manner. Because MLC2 destabilizes cell junctions to increase paracellular flux, the inhibition of Rho/Rho-kinase pathway may improve microvascular endothelial function and suppress barrier permeability.15, 16
In light of the above, the aims of the current study were threefold. First, it was to explore whether PKC-β and RhoA/Rho-kinase/p-MLC2 pathway are involved in HG-mediated cerebral barrier damage using a cell culture model of human BBB. Second, it was to study the nature of correlation between PKC-β and the components of RhoA/Rho-kinase/p-MLC2 pathway in a comprehensive fashion to discover new targets that may be utilized in clinical settings to prevent BBB damage in hyperglycemic ischemic stroke. Finally, it was to assess whether normalization of glucose levels could improve barrier function through modulation of RhoA/Rho-kinase/p-MLC2 pathway, cytoskeleton, or tight junction assembly.
MATERIALS AND METHODS
Cell Culture
Human brain microvascular endothelial cells (HBMEC, >5 × 105 cells) and human astrocytes (>5 × 106 cells) isolated from the cerebral cortex and cryopreserved at passage 1 were purchased from TCS CellWorks (Buckingham, UK). Cells between passages four and seven were cultured in their respective specialized media to ∼90% confluence before exposure to normoglycemia (NG, 5.5 mmol/L
In Vitro Model of Human Blood–Brain Barrier
Human astrocytes were seeded overnight on the outside of polyester membrane (0.4 μm pore size) transwell inserts (Corning Costar, High Wycombe, UK) directed upside down in the culture chamber. On the following day, HBMEC were seeded onto the inner parts of the membranes and cells were grown to confluence.
Blood–Brain Barrier Experiments
Blood–brain barrier integrity was assessed by measurements of transendothelial electrical resistance (TEER) and the flux of Evan's blue-labeled albumin (EBA), a high molecular weight permeability marker (67 kDa), across co-cultures as previously described. 16 Briefly, after measuring TEER across the membranes by STX electrodes and EVOM resistance meter (World Precision Instruments, Hertfordshire, UK), the inserts were transferred to fresh 12-well plates containing 2 mL Hank's buffered salt solution in the basolateral compartments. In the apical chambers, culture media was replaced with 500 μL of Hank's buffered salt solution containing EBA (165 μg/mL). Both luminal and abluminal samples were taken after 1 hour of incubation to determine the optical readings of EBA using a plate reader (absorbance: 610 nm). Flux across cell-free inserts were determined and transport was calculated as ‘cleared volume’ using the following formula. Cleared volume (μL)=concentration(abluminal reading) × volume(abluminal) × concentration(luminal reading)−1.
Immunoblotting
To evaluate the relative differences in protein levels, immunoblottings were performed as previously described. 16
In-Cell Western Analyses
Equal numbers of HBMEC (∼5 × 103) seeded in 96-well plates were subjected to experimental conditions before fixing and permeabilizing them in 3.7% formaldehyde/phosphate-buffered saline and 0.1% Triton X-100/phosphate-buffered saline, respectively. The cells were then successively incubated with primary (5 μg/mL) and infrared dye-tagged species-specific secondary antibodies (Li-Cor Biosciences, Cambridge, UK). The plates were scanned using Odyssey infrared imaging system (Li-Cor BioSciences). Readings representing the aggregate signal of the entire well for target proteins were normalized against those obtained for α-tubulin in the same wells. By simultaneously studying two different targets using spectrally distinct dyes and detection via separate fluorescence detectors and lasers (at 700 and 800 nm), in-cell westerns dramatically enhanced quantification accuracy.
Immunocytochemistry
Human brain microvascular endothelial cells cultured on coverslips were fixed (4% paraformaldehyde/phosphate-buffered saline) and permeabilized (0.1% Triton X-100/phosphate-buffered saline) before visualizing actin cytoskeleton via Rhodamine-labeled phalloidin dye (5 U/mL). To detect RhoA, Rho-kinase, occludin (Santa Cruz Biotech, Dallas, TX, USA) or ZO-1 (Abcam, Cambridge, UK), the cells were successively exposed to the respective primary (overnight at 4°C) and appropriate secondary antibodies. Nuclei were detected by 4,6-diamidino-2-phenylindole staining before viewing cells by fluorescence microscopy.
RhoA Activity Assay
RhoA activity was measured using a commercial kit (Millipore, Billerica, MA, USA). Briefly, HBMEC lysates (∼150 μg) were incubated with Rhotekin Rho-binding peptide immobilized on agarose beads (10 μg). Activated guanosine triphosphate-RhoA bound to Rhotekin was detected by immunoblotting using an anti-RhoA antibody.
Protein Kinase C Activity Assay
Total PKC activity was determined in HBMEC lysates using the PepTag assay kit, which utilizes a brightly colored PKC-specific peptide substrate, PepTag C1 (Promega, Madison, WI, USA). Phosphorylation of peptide substrate by enhanced PKC activity changes the net charge of the peptide from +1 to −1, thereby allowing the separation of phosphorylated and non-phosphorylated peptides via agarose (0.8%) gel electrophoresis. The phosphorylated peptide bands, migrated towards anode, were then excised from the gel under ultraviolet light, melted at 95°C, and solubilized in a mixture containing PepTaq assay gel solubilization solution and glacial acetic acid before reading the absorbances at 570 nm. Total PKC activity (units/mL) was then calculated as per manufacturer's instructions. The specific activity of PKC-β was also measured using the same kit after immunoprecipitation of PKC-β antigen by a specific antibody and Dynabeads protein G (Invitrogen, Paisley, UK).
Small Interfering RNA Knockdown
Human brain microvascular endothelial cells grown to semi-confluence were transfected for 24 hours with DharmaFECT small interfering RNA (siRNA) transfection reagent 4 containing 50 to 100 nmol/L of ON-TARGET plus SMART pool human siRNA against PKC-β and RhoA (Thermo Scientific Dharmacon, Lafayette, CO, USA). Human brain microvascular endothelial cells transfected with non-targeting pool of siRNA served as controls. After 3 days of exposure to experimental conditions, HBMEC were homogenized to assess the levels of protein downregulation by immunoblotting.
Transfection Experiments
Human brain microvascular endothelial cells (5 × 106) were resuspended in 100 μL supplement- and antibiotic-free media and stored on ice in a pre-chilled sterile cuvette. Anti-RhoA IgG (3 μg/cuvette) was added to the cell suspensions before electroporation at 1.8 kV (Electroporator Easyject Prima; Equibio, Ashford, UK). Cells electroporated with equal volumes of vehicle (distilled H2O) containing 3 μg of anti-RhoA IgG served as controls. The viabilities of the electroporated cells were quickly analyzed before plating. As the electroporation process reduced cell viability by ∼39±4%, the number of cells after this procedure was increased by 75% to allow all experimental groups to reach confluence simultaneously. Once the cells are settled, they were subjected to the experimental conditions.
Cell Viability Assay
A small aliquot of cells exposed to different treatment regimens were incubated with 0.1% trypan blue for 5 minutes and viewed under light microscope to determine the level of possible cytotoxicity. The percentage of viable cells was calculated by counting 100 cells.
Statistical Analysis
Data are presented as mean±s.e.m. Statistical analyses were performed using IBM SPSS statistics 20.0 software package. Comparisons of the mean values were carried out by Student's two-tailed t-test or one-way analysis of variance, where appropriate, followed by Dunnett's post hoc testing. P<0.05 was considered significant.
RESULTS
Effects of Hyperglycemia on RhoA/Rho-Kinase/p-MLC2 Pathway Components
Hyperglycemia significantly increased RhoA and Rho-kinase protein expressions, MLC2 mono/di-phosphorylations (p-MLC2Ser19/p-MLC2Thr18-Ser19), and total RhoA activity. These increases were independent of a rise in osmolality and were completely neutralized by normalization of glucose levels. Although suppression of Rho-kinase activity by its specific inhibitor Y-27632 (2.5 μmol/L) did not affect total RhoA protein expression and activity, it led to marked decreases in p-MLC2Ser19 and p-MLC2Thr18-Ser19. Given that Rho-kinase is a downstream effector of RhoA and an upstream mediator of MLC2, these results were somewhat expected. In contrast, electroporation of anti-RhoA IgG into HBMEC successfully neutralized RhoA activity (Figures 1A and C).
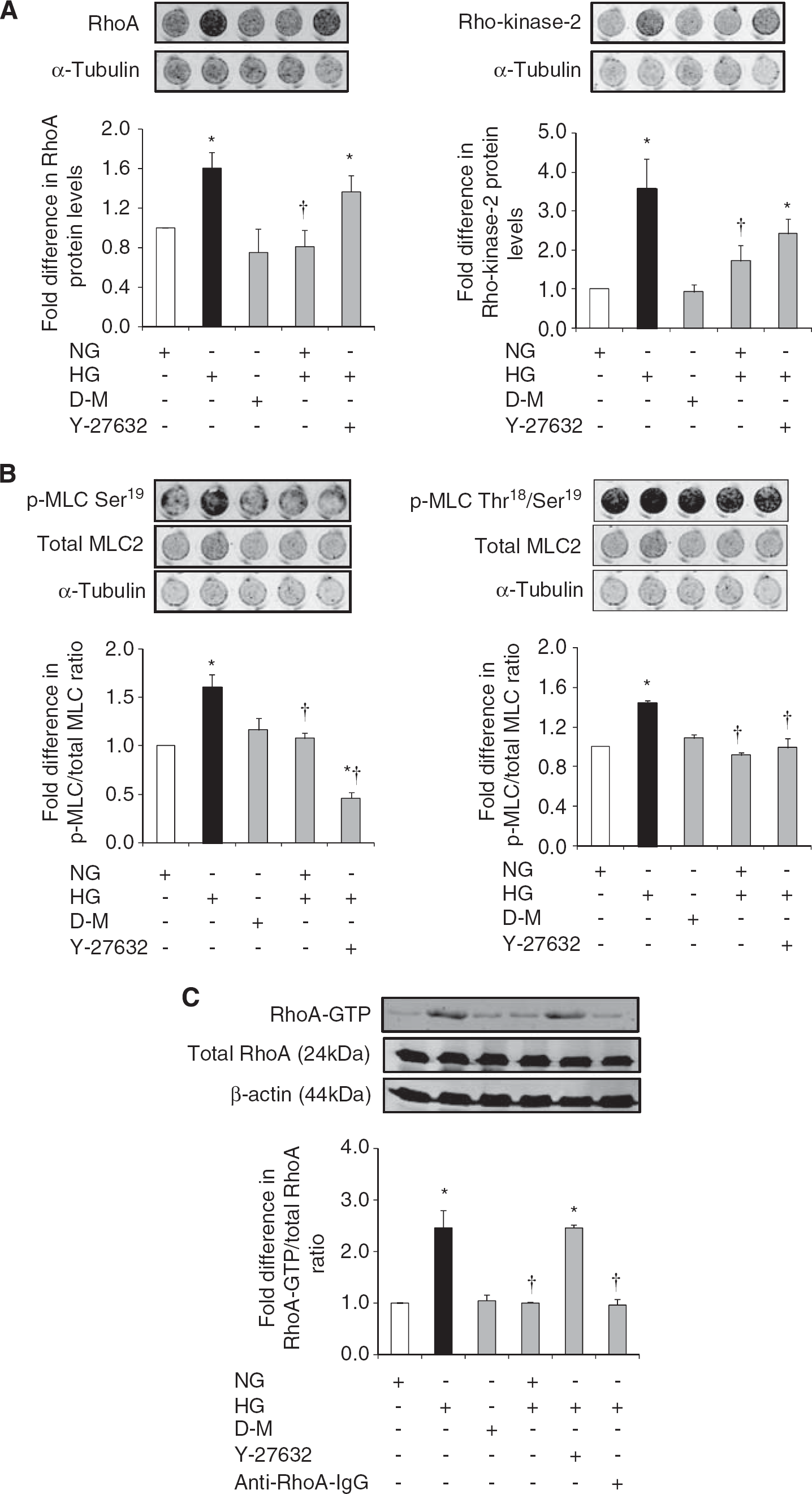
The effects of glucose normalization, Y-27632 and anti-RhoA immunoglobulin G (IgG) on RhoA/Rho-kinase/p-MLC2 pathway components. Representative in-cell western analyses of RhoA and Rho-kinase (
Effects of Hyperglycemia on In Vitro Barrier Integrity and Tight Junction Protein Characteristics
Hyperglycemia compromised in vitro cerebral barrier integrity as evidenced by decreases in TEER and concurrent increases in EBA flux. Normalization of glucose levels and suppressions of RhoA and Rho-kinase activities via electroporation of anti-RhoA IgG and treatments with Y-27632 (2.5 μmol/L), respectively abolished HG-induced barrier dysfunction. Hyperglycemia-mediated decreases observed in occludin levels provided some explanation for the barrier dysfunction. The changes in occludin expression appeared to be independent of an increase in osmolality and were completely prevented by glycemic control and suppression of Rho-kinase activity. Neither HG nor the modulation of glucose levels or Rho-kinase activity had any impact on ZO-1 expression (Figures 2A and D).
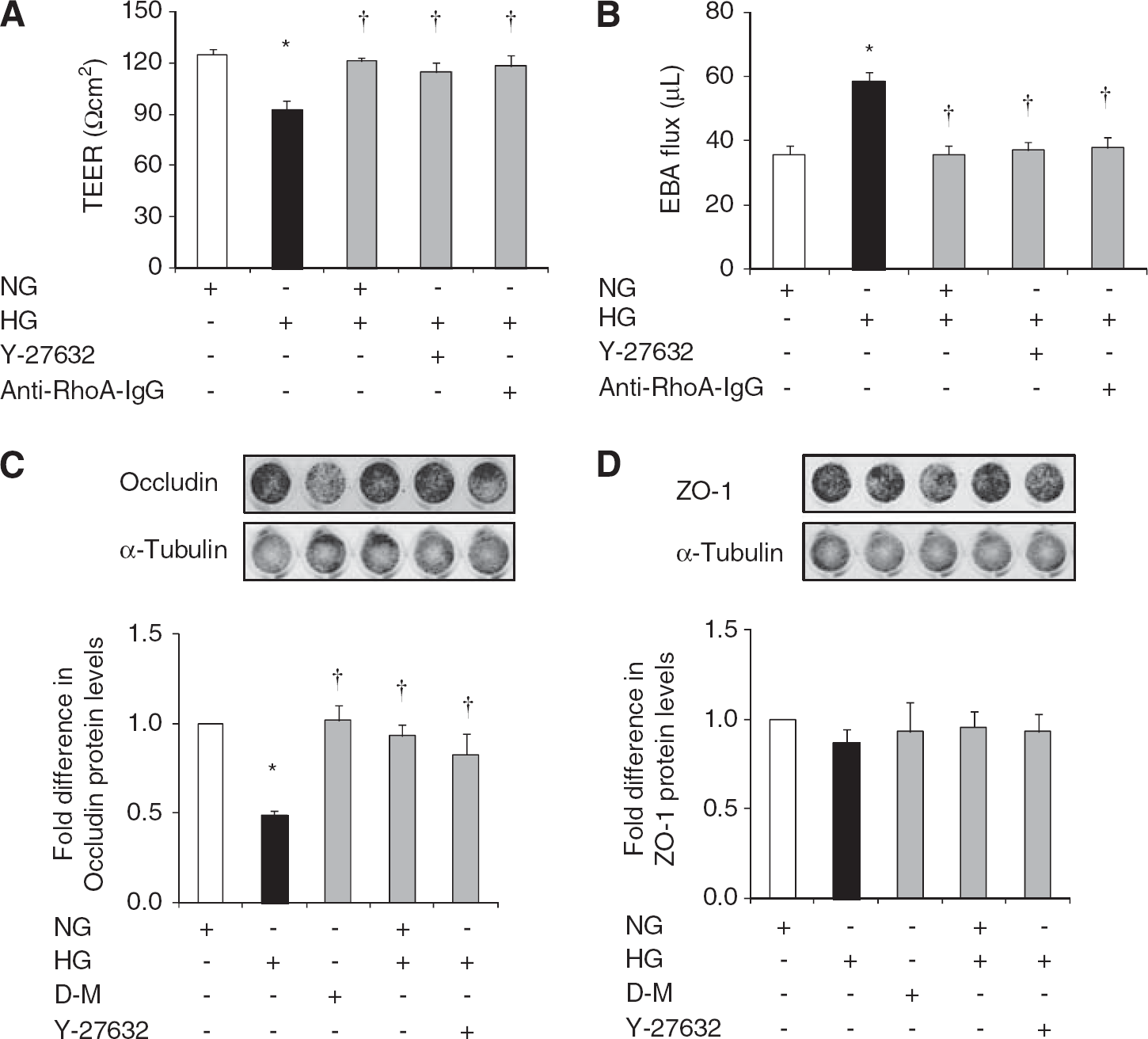
The effects of glucose normalization, Y-27632 and anti-RhoA immunoglobulin G (IgG) on blood–brain barrier (BBB) integrity and tight junction protein expressions. Transendothelial electrical resistance (
Despite failing to alter ZO-1 levels, HG drastically impaired its endothelial cell membrane localization in a similar pattern to that of occludin. Hyperglycemia also promoted endothelial cells stress fiber formation and morphologic changes characterized with their cuboidal or elongated appearance. Normalization of glucose levels and inhibitions of RhoA and Rho-kinase during hyperglycemic insult reinstated cortical actin staining, normal cellular morphology, and peripheral staining of both occludin and ZO-1 (Figures 3A and C).
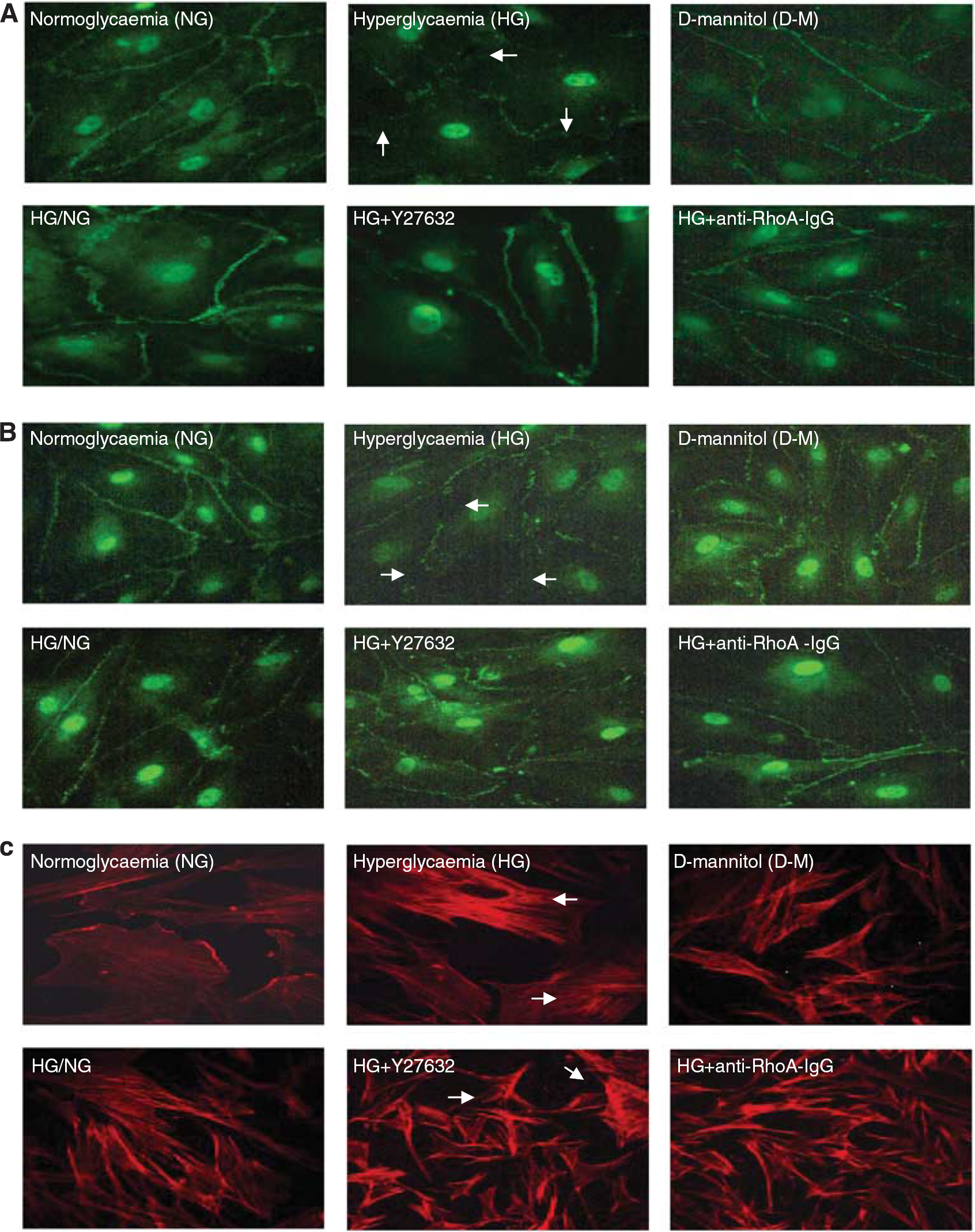
The effects of glucose normalization, Y-27632 and anti-RhoA immunoglobulin G (IgG) on cytoskeleton and tight junction protein localizations. Representative immunocytochemical analyses of occludin (
Effects of Hyperglycemia on Protein Kinase C-β Activity and Associated Downstream Mediators
Hyperglycemia significantly increased total PKC activity in HBMEC. Exposure of hyperglycemic cells to LY333531 (5 μmol/L), a selective PKC-β inhibitor, diminished the level of total PKC activity below the levels detected in normoglycemic conditions and proved the efficacy of LY333531 in suppressing both inducible and constitutive PKC activities. Inhibition of PKC-β also improved barrier integrity and function as confirmed by TEER and paracellular flux studies, respectively (Figure 4A).
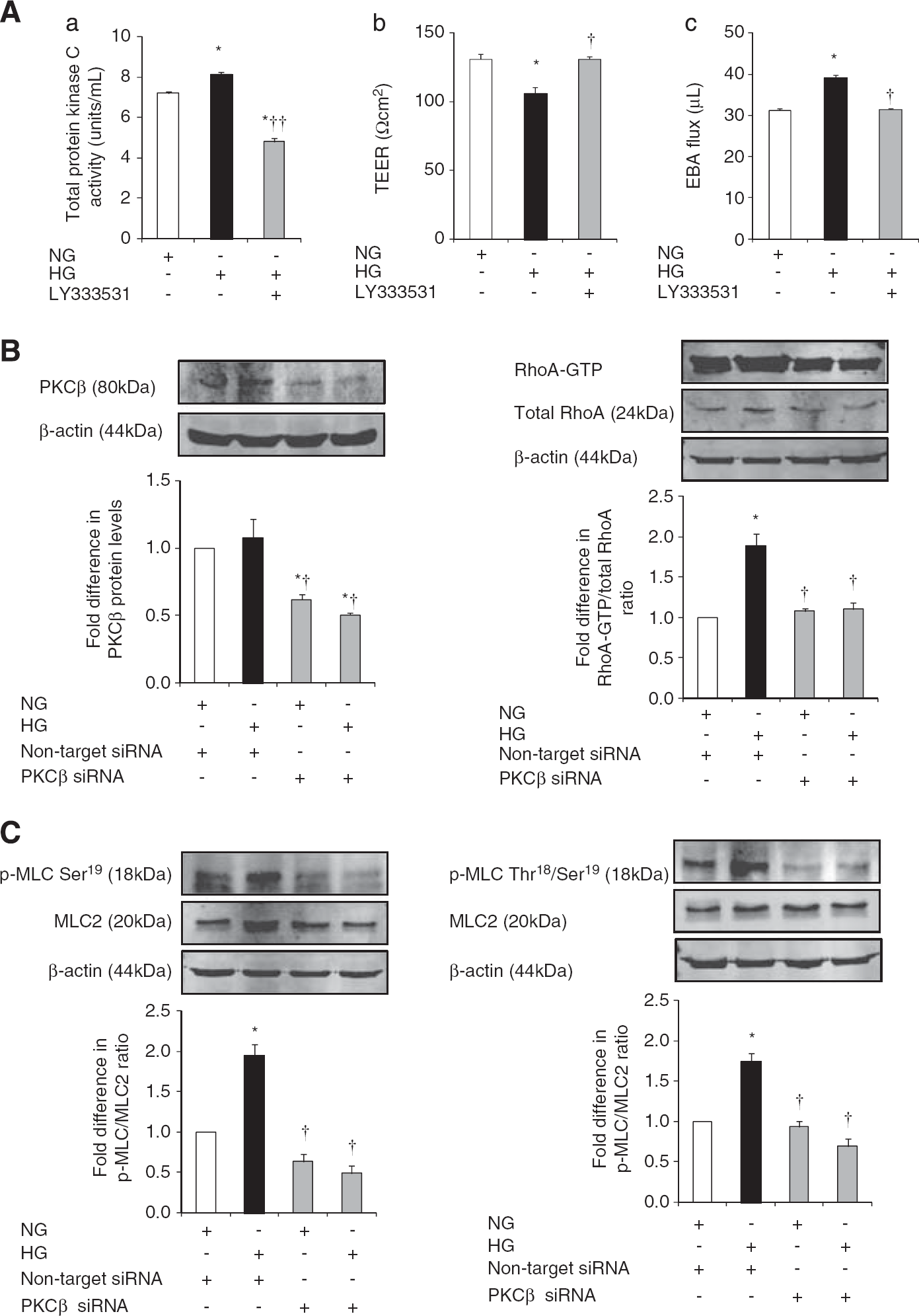
The effects of inhibition of protein kinase C-β (PKC-β) activity on blood–brain activity (BBB) integrity, RhoA activity and p-MLC2. (
Attenuation of PKC-β activity via specific siRNA neutralized HG-evoked increases in RhoA activity, p-MLC2Ser19 and p-MLC2Thr18-Ser19 and thus proved the direct involvement of this pathway in PKC-β-induced cerebral barrier damage (Figures 4B and C).
Effects of Protein Kinase C-β Inhibition on Tight Junctions and Cytoskeleton
As mentioned above, HG evoked a significant decrease in HBMEC occludin protein expression without affecting that of ZO-1. Small interfering RNA-mediated attenuation of PKC-β activity dramatically increased occludin and ZO-1 expressions under hyperglycemic conditions and produced a selective increase in occludin expression under normoglycemic conditions. Similarly, inhibition of PKC-β activity preserved cortical actin staining and normal cellular structure in cells subjected to HG (Figures 5A and C).
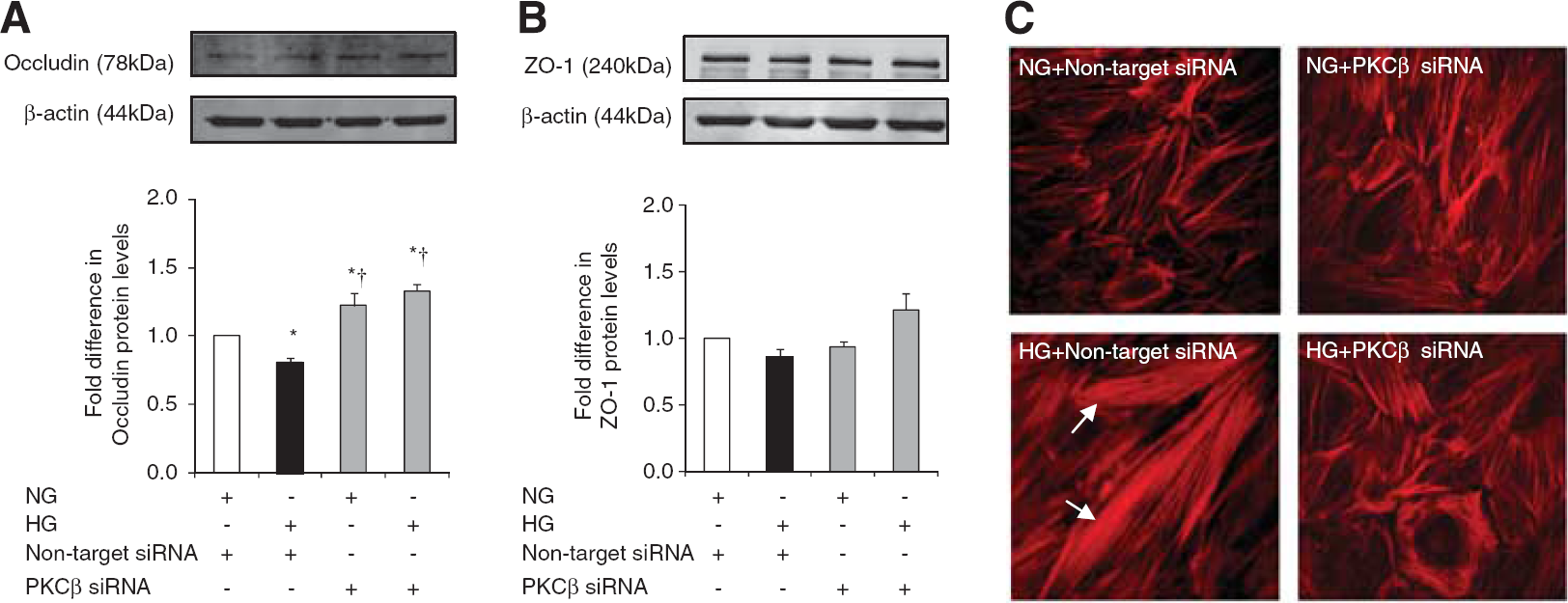
The effects of protein kinase C-β (PKC-β) activity inhibition on tight junction protein levels and cytoskeleton. Occludin (
RhoA and Rho-Kinase Regulate Protein Kinase C Activity in a Reciprocal Manner
Suppression of RhoA protein levels in HBMEC abolished HG-mediated increases in PKC-β activity. Furthermore, inhibitions of RhoA and Rho-kinase activities by electroporation of anti-RhoA IgG and Y-27632 (2.5 μmol/L), respectively markedly decreased total PKC activity. Taken together, these findings support counterregulatory effects of RhoA and Rho-kinase on PKC-β activity (Figures 6A and C).
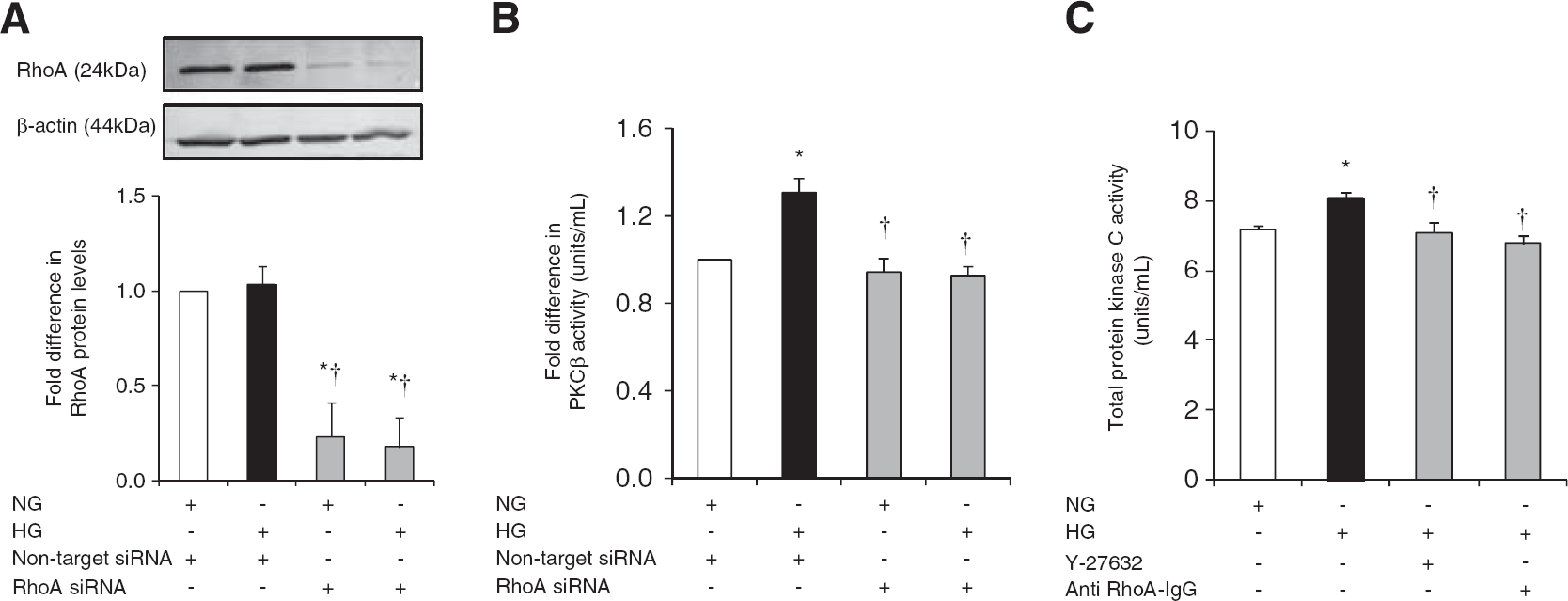
The impact of RhoA activity suppression on total protein kinase C (PKC) and PKC-β activities. Representative immunoblotting of RhoA expression (
DISCUSSION
Brain edema associated with increased rates of patients' mortality appears to be more prevalent in stroke patients with diabetes or stress HG than those without thereby implying a prominent role for HG in the BBB breakdown.5, 6 Although HG represents a common pathology in ischemic stroke patients, the mechanisms by which it may compromise the cerebral barrier integrity and/or exacerbate vascular damage remain largely unknown. Bearing these in mind, the current study focused on the relevance of two apparently distinct pathways, namely PKC and RhoA/Rho-kinase/MLC2 to HG-mediated barrier damage using an in vitro model of human BBB composed of HBMEC and human astrocytes. It is thought that elucidating the nature of the correlation between these pathways that collectively regulate vasomotor function, cell proliferation, survival, migration, oxidative stress, and immune responses may prove extremely beneficial in identifying novel targets for future therapeutic approaches.17, 18
Exposure of HBMEC to HG led to significant increases in RhoA activity and RhoA and Rho-kinase protein expressions, which were fully mitigated by normalization of glucose levels. Although clinical evidence regarding the necessity and extent of glycemic control during acute ischemic stroke is debatable, it is important to monitor and possibly reduce blood glucose levels in an effective manner considering the close correlation between HG and poor outcomes, typically defined by enhanced mortality or dependence 90 days after a stroke.5, 6 As equimolar concentrations of
Hyperglycemia severely perturbed the cerebral barrier integrity and function in in vitro settings as ascertained by marked decreases in TEER and concomitant increases in EBA flux, respectively. Normalization of glucose levels and inhibitions of RhoA and Rho-kinase eradicated the deleterious effects of HG on the in vitro barrier. Similar to RhoA activity, HG also significantly elevated total PKC and PKC-β activities, which appear to require unabated activities of RhoA and Rho-kinase. Subsequent studies, through assessments of TEER and EBA flux, proved the regulatory function of PKC-β in maintaining the cerebral barrier integrity and function and affirmed the findings of recent studies in which the inhibition of PKC-β substantially improved mouse brain microvascular endothelial cell and human umbilical vein endothelial cell barrier integrities by augmenting cellular anti-oxidant and anti-apoptotic capacities.11, 22 Additional studies designed to assess the specific impacts of PKC-β on RhoA/Rho-kinase/p-MLC2 complex demonstrated dramatic reductions in RhoA and Rho-kinase protein expressions and p-MLC2 in PKC-β-knocked down HBMEC subjected to HG.
The inhibitory reciprocal relationship between total PKC/PKC-β and RhoA activities in hyperglycemic settings necessitated a closer scrutiny of the correlation between RhoA and PKC-β to reveal the true nature of HG-mediated pathologic cascade resulting in the breakdown of cerebral barrier. Analyses of PKC-β activity in HBMEC transfected with specific RhoA siRNA led to selective decreases in enzyme activity and further substantiated the counterregulatory effect of RhoA on PKC-β activity.
Changes in cellular architecture may also contribute to HG-induced barrier failure. Indeed, experiments examining the structural differences in microfilaments, a crucial element of cytoskeleton, showed that HBMEC grown under normal conditions displayed a cortical actin staining whereas those subjected to HG possessed actin stress fibers traversing the cells and took up predominantly elongated and somewhat cuboidal appearance. The appearance of stress fibers coincided with enhanced MLC2 phosphorylation at Ser19 (p-MLC2Ser19) and Thr18-Ser19 (p-MLC2Thr18-Ser19) residues possibly through activations of MLC kinase and Rho-kinase and/or inhibition of MLC phosphatase. Once established, stress fibers create a tensile centripetal force to pull TJ proteins inward to compromise junctional integrity and widen intercellular gaps. 16 RhoA/Rho-kinase pathway may also modulate actin filament (de)stabilization by phosphorylating adducin, LIM kinase, and ezrin-radixin-moesin proteins, which promote the binding of cytoskeletal proteins, e.g., spectrin and cofilin to cortical actin.23, 24 In addition to stress fiber formation, the activation of RhoA/Rho-kinase pathway may also disrupt the BBB integrity through attenuations of endothelial nitric oxide synthase expression, phosphorylation (at Ser1177), and activity and consequently deplete the availability of barrier-protective nitric oxide. Indeed, suppression of RhoA activity by statins and greater availability of nitric oxide have been shown to produce the opposite effects. 25 Similarly, the inhibitions of RhoA and Rho-kinase before and during hyperglycemic challenge as well as normalization of glucose levels in the current study has also led to normalization of p-MLC2, prevention of stress fiber formation, restoration of actin staining to cell periphery, and abolishment of the barrier-disruptive effects of HG. In concert with its barrier-protective effects, the stress fiber formation was fully mitigated by PKC-β inhibition.
Other mechanisms including overt breakings in interendothelial junctional complex may also promote the cerebral barrier failure in hyperglycemic settings. The reduced expression of TJ protein occludin may shed some light on the HG-induced barrier hyperpermeability and support the previous in vivo studies reporting a direct correlation between the levels of TJ proteins and the extent of endothelial barrier permeability.26, 27, 28 The discontinuous appearances of occludin and ZO-1 on cell periphery provide further explanation for the HG-mediated increases in paracellular flux. Consistent with the recent data showing an insulin-mediated elevation in the cerebral contents of occludin and ZO-1, normalization of glucose levels in our study also significantly improved occludin levels and effectively restored occludin and ZO-1 staining to the cell periphery, a set of findings replicated by inhibitions of RhoA or Rho-kinase.26, 27, 28 Subsequent studies focusing on the specific impact of PKC-β on cerebral endothelial cell content of TJ proteins have shown that knockdown of PKC-β significantly increased occludin and ZO-1 protein levels under hyperglycemic conditions and selectively that of occludin also under normoglycemic conditions. Decreases in phosphorylation, ubiquitination, and trafficking of occludin may account for the PKC-β-inhibition-mediated improvements observed in occludin protein levels. 29 Likewise, decreases in occludin and claudin-5, another TJ protein, phosphorylation may also account for the barrier-protective effects obtained with inhibition of endothelial Rho-kinase activity. 30
Although not investigated in the present study, it is likely that HG may also affect the expression and localization of claudin-5, a key component of the BBB known to prevent interendothelial cell leakage of small molecules (<800 kDa). 31 In support of this notion, using an in vitro model of human BBB comprising HBMEC, human astrocytes, and human pericytes, a recent study has shown that HG markedly increases the paracellular flux of sodium fluorescein (376 kDa) through a complex mechanism involving activations of several PKC isoforms, namely PKC-α, β, or βII. 32 Bearing HG-induced cytoskeletal reorganization in mind, possible changes exerted on HBMEC-matrix interactions regulated via β1-integrin may also contribute to barrier permeability by directly affecting claudin-5 expression as recently reported. 33
In conclusion, the current study demonstrates that activations of PKC-β or RhoA/Rho-kinase/p-MLC2 pathway under hyperglycemic conditions mediate the structural and functional impairment of an in vitro model of human cerebral barrier. Considering the regulatory effects of PKC-β on the expression, phosphorylation, and/or activity of RhoA/Rho-kinase/p-MLC2 pathway components and organization of cytoskeleton and TJ assembly, it is plausible to suggest that PKC-β may be an efficacious novel therapeutic target to stem the cerebral barrier dysfunction in hyperglycemic stroke.
Despite existence of a recent study showing that the inhibition of PKC-βII by CGP53353 reverses enhanced BBB permeability and prevents edema formation in streptozotocin-induced diabetic rats subjected to middle cerebral artery occlusion, 34 it remains essential to extend our study to in vivo settings by using a well-established animal model of focal cerebral ischemia with/out diabetes. This is particularly important given the fact that much of the existing animal data with hyperglycemic focal ischemia, although invaluable, are somewhat irrelevant to clinical scenario because of the choice of wrong animal models, excessive blood glucose levels, and inconsistent findings with insulin. 35 For instance, although the majority of stroke patients have unrecognized insulin resistance coupled with type-2 diabetes, in most of the experimental studies a streptozotocin-induced model of type-1 diabetes is used. 35 Other than being unrepresentative of >90% of clinical cases, streptozotocin has been shown to impair brain microvasculature and increase apoptosis in middle cerebral artery occlusion-induced models of focal cerebral ischemia. 36
There are some limitations to the current study. First, a single marker of paracellular transport, i.e., EBA (67 kDa) has been employed throughout this study. The use of fluorescent-labeled Dextran of higher molecular weights may have shed some light on the size of intercellular openings. Furthermore, using a cell culture model of human BBB established with relatively late passage endothelial cells and astrocytes, in this study only one factor, i.e., glucose, out of many involved in ischemic damage could be manipulated, whereas a number of pro-inflammatory cytokines and circulating components during an ischemic stroke, notably tumor necrosis factor-α, interleukin-6, vascular endothelial growth factor, and thrombin, are known to affect BBB permeability.37, 38
Footnotes
The authors declare no conflict of interest.