
Abstract
Ischemic preconditioning (PC) can markedly reduce ensuing ischemic damage. Although most attention has focused on the neuronal effects of PC, the authors have recently shown that ischemic PC reduces ischemia-induced cerebrovascular damage. In vivo, it is difficult to ascertain whether this is a direct cerebrovascular effect of PC. This study, therefore, examined whether cerebral endothelial cells can be preconditioned in vitro in the absence of other cell types. Experiments were performed on an immortalized mouse brain endothelial cell line or primary cultures of mouse brain microvessel endothelial cells. Cells were exposed to oxygen glucose deprivation (OGD) of either short duration, as a PC stimulus, or a long duration (5 hours) with or without reoxygenation to induce endothelial damage. Endothelial injury was assessed by measuring lactate dehydrogenase release and the expression of intercellular adhesion molecule-1 at the protein and mRNA levels. Experiments indicated that 1 hour of OGD was the optimal PC stimuli and that a 1 or 3 day interval was the optimal time interval between the PC stimulus and the injurious event. Preconditioned cells had less lactate dehydrogenase release during OGD (± reoxygenation) and reduced intercellular adhesion molecule-1 expression after OGD with reoxygenation. This study shows that cerebral endothelial cells can be directly preconditioned. The importance of this phenomenon in the overall effects of PC on the brain remains to be elucidated. Understanding the protective mechanisms elicited by PC may give insight into how to prevent ischemia-induced vascular damage (e.g., hemorrhagic transformation).
Ischemic preconditioning (PC) was first described in the heart (Murry et al., 1986). In that tissue, repeated short periods of ischemia protect against a subsequent long duration of ischemia with reperfusion. Subsequent studies have shown that the brain can also be preconditioned to reduce ischemic brain damage. The ischemic PC stimuli can be either a short period of global ischemia (Kato et al., 1991; Kitagawa et al., 1990) or short period(s) of focal cerebral ischemia (Barone et al., 1998; Chen et al., 1996). Ischemic PC can be demonstrated both in vivo and in neuronal cells cultures, and that has allowed the use of such cultures to examine the mechanisms involved in PC (Dawson and Dawson, 2000; Gonzalez-Zulueta et al., 2000).
Whether other brain cell types and, in particular, cerebral endothelial cells can also be preconditioned has received very little attention. We have recently shown in vivo that ischemic preconditioning reduces the blood–brain barrier disruption and brain edema formation that follows subsequent focal cerebral ischemia in the rat (Masada et al., 2001). Preconditioning was also associated with less induction of heat shock protein 70, a stress marker, in the cerebrovasculature (Masada et al., 2001). These results indicate that PC can protect the cerebrovasculature. However, in vivo, it is difficult to prove that this is a direct effect rather than the result of less parenchymal cell damage. The current study, therefore, examined whether PC can protect cerebral endothelial cells in vitro.
MATERIALS AND METHODS
This study comprised four sets of experiments. In all the experiments, cerebral endothelial cells were exposed to a short duration of oxygen-glucose deprivation (OGD) as a PC stimulus. After an interval, they were then exposed to a longer duration of OGD (5 hours) with or without reoxygenation to examine whether prior PC would reduce injury. The first set of experiments examined the optimal duration of the PC stimulus using an immortalized mouse brain endothelial cell line (bEND.3). In the second set of experiments, the duration of the PC stimulus was kept constant, but the time interval between that stimulus and the prolonged OGD was varied. The third set of experiments compared the effects of PC on two types of cerebral endothelial cells, bEND.3 and primary cultures of mouse brain microvessel endothelial cells (mBMEC). For all three of these sets of experiments, lactate dehydrogenase (LDH) release to the media was used to assess endothelial injury. The fourth set of experiments examined the effect of PC on the upregulation of endothelial (bEND.3) intercellular adhesion molecule (ICAM)-1 mRNA and protein levels that occurs after OGD with reoxygenation.
Mouse cerebral endothelial cells
For mBMEC preparation, 4- to 6-week-old CD-1 mice were used. Briefly, the brain was minced in Hanks balanced salt solution (Invitrogen CA, U.S.A.) and homogenized gently in Dounce type homogenizer. The microvessels were cleaned from myelin using a 13% Dextran solution (USB, OH, U.S.A.), and separated from erythrocytes using a Percoll (Pharmacia, NJ, U.S.A.) gradient. The microvessels were digested in Hanks balanced salt solution containing 1-mg/mL collagenase/dispase (Roche, IN, U.S.A.) for 40 minutes at 37°C. BMEC were cultured in Dulbecco's Modified Eagle Medium (DMEM, Invitrogen) supplemented with 10% inactivated fetal calf serum (Invitrogen), 2.5-μg/mL heparin (Sigma, St. Louis, MO, U.S.A.), 20-mmol/L HEPES, 2-mmol/L glutamine and antibiotic/antimycotic (Invitrogen), endothelial cell growth supplement (BD Bioscience, NJ, U.S.A.) and grown in six-well plates coated with collagen IV (BD Bioscience, KY, U.S.A.).
Brain endothelial cell line (bEnd.3) was purchased from ATCC (American Type Culture Collection, VA, U.S.A.). Cells were plated and grown in the recommended cell media (DMEM, 10% fetal bovine serum, 1 × AA, 2-mmol/L glutamine) in the presence of 10% CO2.
Oxygen glucose deprivation
Confluent mouse brain endothelial cell cultures (mBMEC and bEnd.3) were transferred into a temperature-controlled (37° ± 1°C) anaerobic chamber (Coy Laboratory, MI, U.S.A.) containing a gas mixture composed of 5% CO2, 10% H2, 85% N2. This chamber maintains a strict 0 to 5 ppm oxygen atmosphere through the hydrogen gas reacting with a palladium catalyst to remove oxygen. The culture medium was replaced with deoxygenated glucose-free DMEM solution, and cells were maintained in the anaerobic chamber for up to 5 hours. Cells were exposed to 15, 30, 45, or 60 minutes of oxygen glucose deprivation (OGD) for preconditioning and for 5 hours to induce injury. Control cell cultures were not exposed to OGD. The anaerobic chamber allowed serial media sampling while the cells were still in the chamber. Sampled media replaced with the same amount of deoxygenated glucose-free DMEM solution. In some experiments, cells that underwent OGD were returned to a normoxic incubator under 5% CO2/95% air for up to 72 hours (reoxygenation).
Lactate dehydrogenase assay
Lactate dehydrogenase (LDH) activity in cell culture media (supernatant) was measured using a commercially available kit, CytoTox 96 (Promega, Madison, WI, U.S.A.), according to the manufacturer's instructions.
Cell viability assay
Cell viability was assessed using a commercial kit (LIVE/DEAD Viability/Cytotoxicity assay kit, Molecular Probes) according to the manufacturer's instructions. The assay for live cells uses calcein-AM, a nonfluorescent esterase substrate, which inside living cells is hydrolyzed by intracellular esterases to calcein with bright-green fluorescence at excitation/emission wavelengths of 495/515 nm. The assay for dead cells uses ethidium homodimer-1 (EthD-1), a molecule that binds to DNA and then fluoresces red at excitation-emission wavelengths of 495/635 nm. Briefly the samples were washed 3 times in Dulbecco's phosphate-buffered saline (Invitrogen), and then were incubated with 2-μmol/L calcein-AM and 4-μmol/L EthD-1 in Dulbecco's phosphate-buffered saline for 45 minutes at room temperature. The buffer was then replaced and left for additional 1 hour. Every sample was done in triplicate. For “dead controls,” cells were fixed with 4% paraformaldehyde for 20 minutes before staining with calcein-AM and EthD-1. All samples were viewed on the confocal microscope Zeiss 510. For every sample, 10 randomly selected spots were captured. Quantification of dead cells (percent of dead cells) was performed using a 1.62 NIH image software system.
In addition, the morphology of cells subjected to OGD and reoxygenation with and without preconditioning was examined using phase contrast (Olympus CK2 inverted microscope).
Western blotting
Samples were lysed with RIPA buffer (10-mmol/L TRIS, 140-mmol/L NaCl, 1% Triton, 1% Na-deoxycholate, 0.1% SDS, 0.5-mmol/L phenylmethylsulfonyl fluoride and supplemented with cocktail inhibitors; Roche). Extracts were homogenized and insoluble debris removed by centrifugation at 6,000 g for 10 minutes at 4°C. Protein concentration in the resulting supernatants was calculated using a Pierce protein assay kit according to the manufacturers instructions (Pierce, IL, U.S.A.). Equal amounts of protein samples (15 μg) were loaded, separated using 12% SDS-polyacrylamide gel electrophoresis and then transferred to Trans-Blot nitrocellulose membrane (BioRad, CA, U.S.A.). The membrane was blocked with a 5% solution of nonfat milk in TBS, for 1 h at room temperature and then incubated with goat anti-mouse ICAM-1 antibody for 18 hours at 4°C. After washing three times with 0.05% (vol/vol) Tween-20 solution in TBS, the membrane was incubated with anti–goat-HRP conjugated antibody (Vector Laboratories, CA, U.S.A.) for 2 h and washed three times with 0.05% Tween-20 in TBS. Chemiluminescent reaction was developed using a chemiluminescent HRP substrate kit (Pierce).
Semiquantitative reverse-transcription polymerase chain reaction
Semiquantitative reverse-transcription polymerase chain reaction was performed to quantify levels of ICAM-1 mRNA expression. Total RNA was prepared from the cultures (1 × 106 cells) using the RNAgents, Total RNA Isolation System (Promega) according to the manufacturer's instructions. Aliquots (3 μg) of RNA were reverse transcribed to cDNA using the Gibco BRL cDNA synthesis system (Gibco BRL, MD, U.S.A.). The following ICM-1 primers were used: 5'TGCGTTTTGGAGCTAGCGGACCA3' and 5'CGAGGACCATACAGCACGTGCAG3'. For β-actin, commercially available primers were used (Biosource International, CA, U.S.A.). The PCR cycles included general denaturation at 94°C (1 minute), annealing at 55°C (1 minute) and extension at 72°C (1 minute), except for the first cycle, which had 4-minute denaturation, and the last cycle, which had 10-minute elongation. A total of 40 amplification cycles for ICAM-1 and β-actin were applied. The PCR products were resolved using electrophoresis in a 2% agarose gel in 1× TBE buffer (Tris-HCl/EDTA /boric acid, pH 8). The gel was stained with ethidium bromide and photographed. The relative densities/volumes of the bands on the film negatives were measured using the NIH image software package.
Statistics
Experiments were performed on at least three separate cultures unless stated, with LDH measurements on each culture being performed in triplicate. Values are expressed as means ± SD. Differences between experimental groups were analyzed by analysis of variance with a Dunnett test for multiple comparisons to a single control group.
RESULTS
Preconditioning bEND.3 cells with either 15, 30, 45, or 60 minutes of OGD 24 hours previously protected the cells from the injury induced by 5 hours of OGD with or without reoxygenation as assessed by LDH release (Fig. 1A and Fig. 1B). The degree of protection increased with the duration of the PC stimuli, although 45 and 60 minutes of PC produced approximately equal protection. Effects of PC on LDH release were mirrored by morphologic changes in the endothelial monolayers (Fig. 1C).
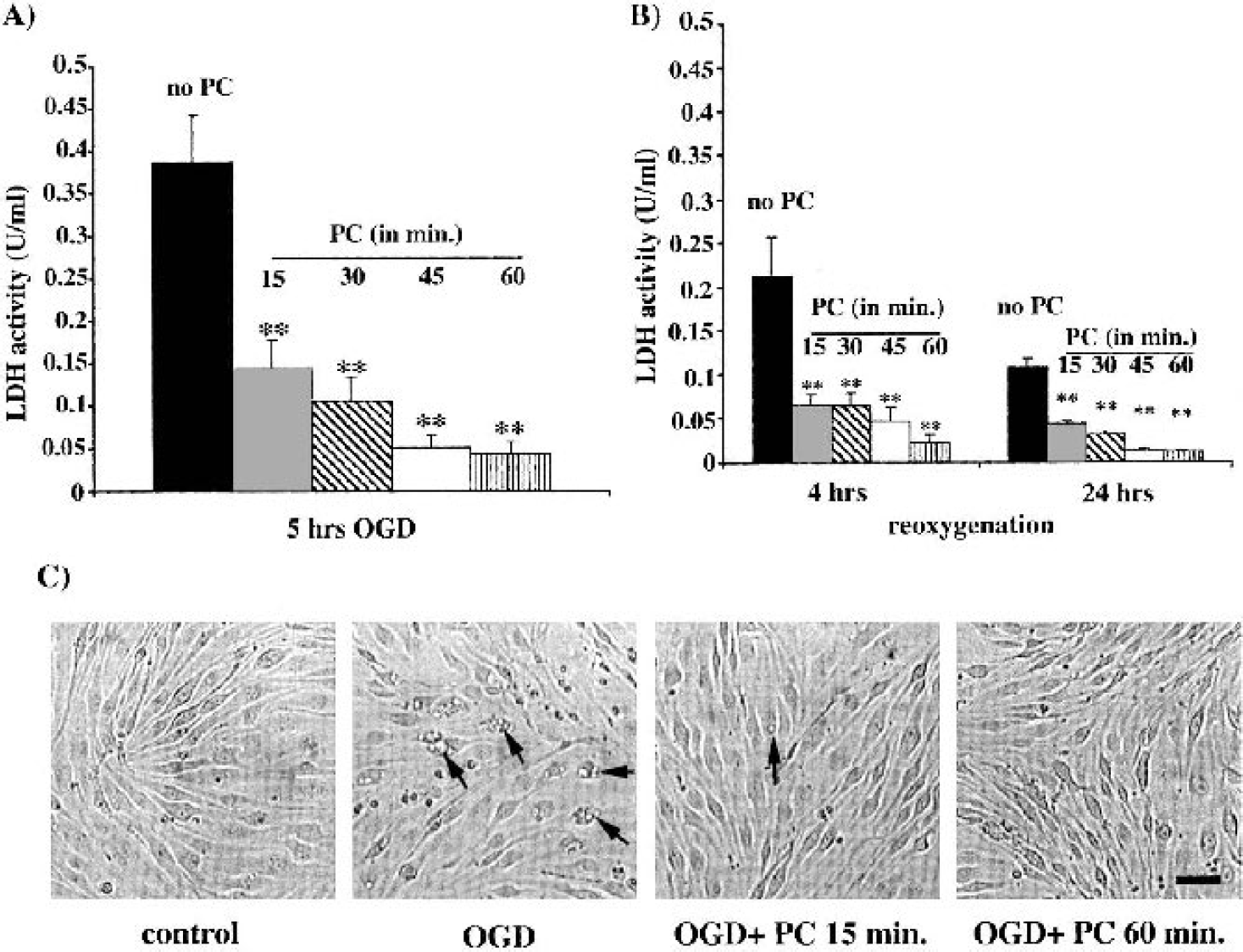
(
Using 1 hour of OGD as the PC stimuli, the effect of altering the time interval between the PC stimuli and the injury inducing OGD was examined in bEND.3 cells. No effect on LDH release was found with an interval of 4 hours, and although some protection was found at 12 hours, maximal protection was found at 24, 48, and 72 hours (Fig. 2). To investigate whether there might be a biphasic PC effect, another set of experiments examined whether shorter intervals might also be protective. Preconditioning 0.5, 1, 2, 3, and 4 hours before 5 hours of OGD and 24 hours of reoxygenation did not reduce LDH release (Fig. 3). Indeed, at the 0.5-hour interval, PC significantly elevated LDH release.
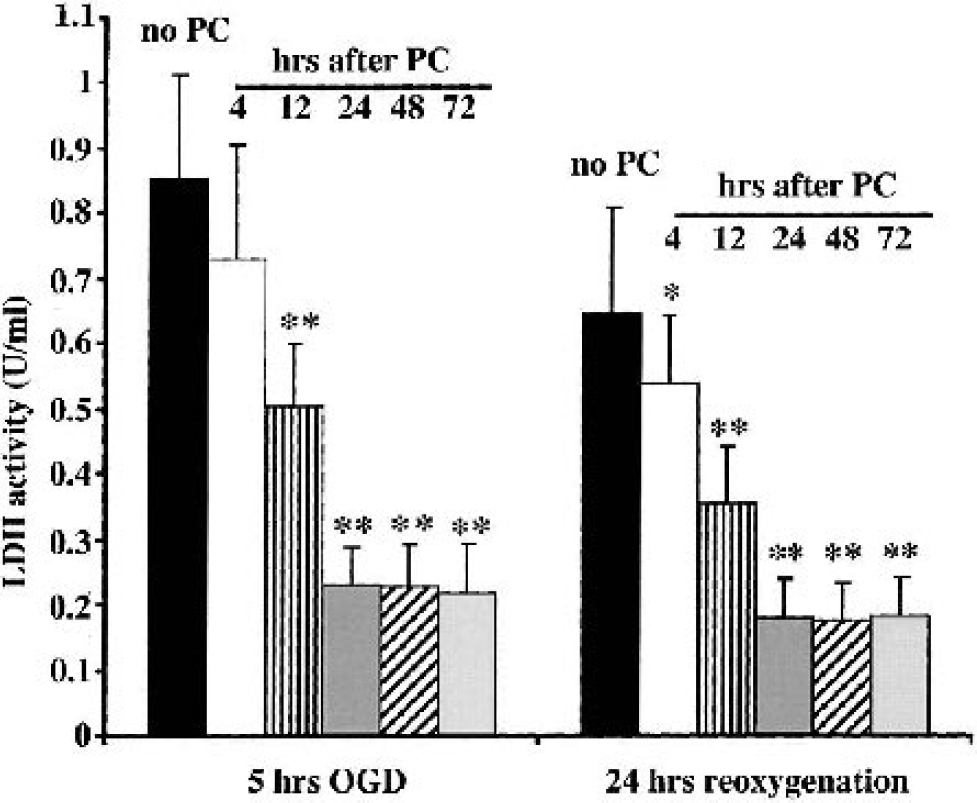
Effects of altering the time interval between the PC stimuli and injury inducing OGD on media LDH activity. bEND.3 cells were exposed to 1 hour of OGD (the PC stimuli) and then 4, 12, 24, 48, or 72 hours later they were exposed to 5 hours of OGD and 24 hours of reoxygenation. Results were compared to cells that were exposed to OGD and 24 hours of reoxygenation without preconditioning (No PC). Media was sampled for measurement of LDH activity at the end of the 5 hours of OGD or after 24 hours of reoxygenation. The cells were placed in fresh media after sampling. Values are mean ± SD; n = 3 separate cultures. Asterisks indicate significant differences from the nonpreconditioned group at the ∗ P < 0.05 and ∗∗ P < 0.01 levels.
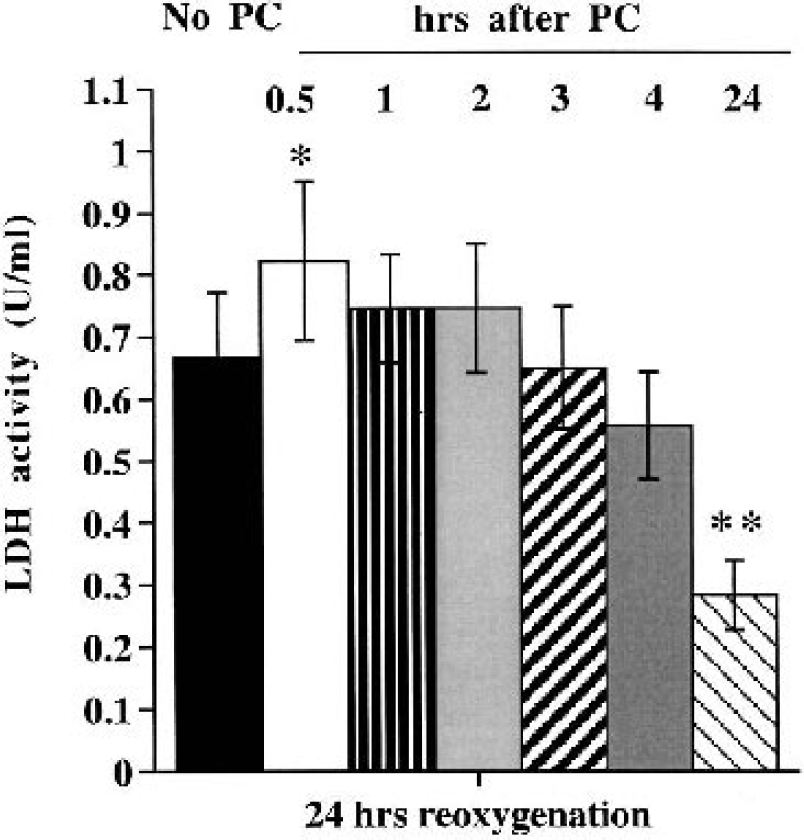
Effects of short time intervals between the PC stimuli and injury inducing OGD on media LDH activity. bEND.3 cells were exposed to 1 hour of OGD (the PC stimuli) and then 0.5, 1, 2, 3, 4, or 24 hours later they were exposed to 5 hours of OGD and 24 hours of reoxygenation. Results were compared with cells that were exposed to OGD and 24 hours of reoxygenation without preconditioning (No PC). Media was sampled for measurement of LDH activity after 24 hours of reoxygenation. Values are mean ± SD; n = 3 separate cultures. Asterisks indicate significant differences from the non-preconditioned group at the ∗ P < 0.05 and ∗∗ P < 0.01 levels.
With an hour PC stimulus and a 24-hour interval between the PC stimulus and the injury inducing OGD, we confirmed the protective effects of PC in bEND3 cells using a viability assay (using calcein-AM and EthD-1). In the absence of PC, 5 hours of OGD resulted in about 20% cell death, and this number increased to about 40% by 24 hours of reoxygenation. Preconditioning significantly reduced this cell mortality (Fig. 4).
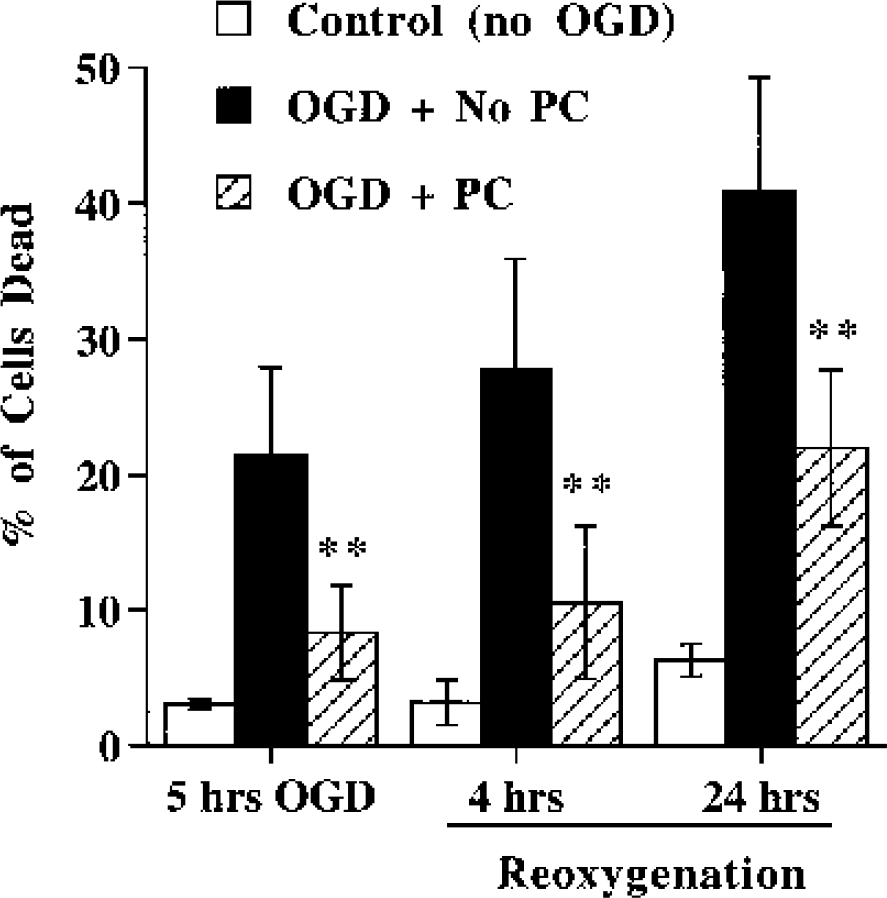
Effects of PC on cell viability after exposure to OGD. bEND.3 cells were preexposed to 1 hour of OGD (the PC stimuli) or no OGD. Twenty-four hours later the cells were exposed to 5 hours of OGD and 0, 4, or 24 hours of reoxygenation. Cell viability was then examined with a calcein-AM and EthD-1 assay, and the percentage of dead cells was determined. In contrast to control cells (not subjected to PC or 5 hours of OGD), which showed a low percentage of dead cells, there was progressive cell death in the No PC group exposed to 5-hour OGD and reoxygenation. This cell death was significantly reduced in the PC group. Values are mean ± SD; n = 3 separate cultures. Asterisks indicate a significant difference from the nonpreconditioned group at the P < 0.01 level.
Because of these initial results on bEND.3 cells, the next set of experiments comparing the effects of PC in mBMEC and bEND.3cells used 1 hour as the PC stimuli and 24 hours as the time interval between PC and the 5 hours of OGD. Both during the 5 hours of OGD (Fig. 5) and during 24 hours of reoxygenation (Fig. 6), PC reduced LDH release in both cell types. The degree of protection was very similar in both cell types; i.e., it was not model dependent.
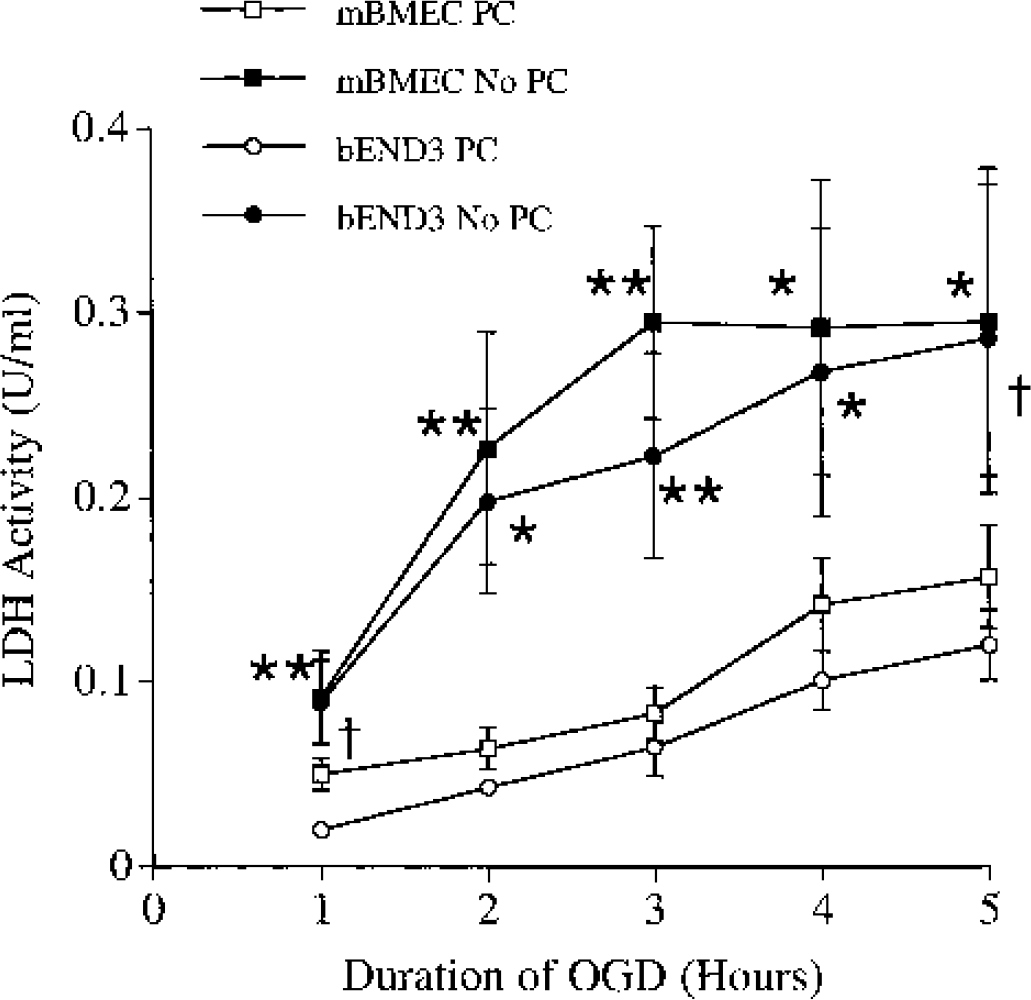
Effect of PC on media LDH activity in bEND.3 cells and mBMEC exposed to 5 hours of OGD. bEND.3 cells and mBMEC were exposed to 1 hour of OGD (the PC stimuli) 1 day before exposure to 5 hours of OGD. The medium was sampled for LDH activity measurements at 1-hour intervals. Results were compared with cells of each type that had not been preconditioned (No PC). Values are mean ± SD; n = 3 separate cultures. Asterisks indicate significant differences from the corresponding No PC group at the ∗ P < 0.05 and ∗∗ P < 0.01 (two-tailed) levels; dagger indicates a significant difference at the P < 0.05 (one-tailed) level.
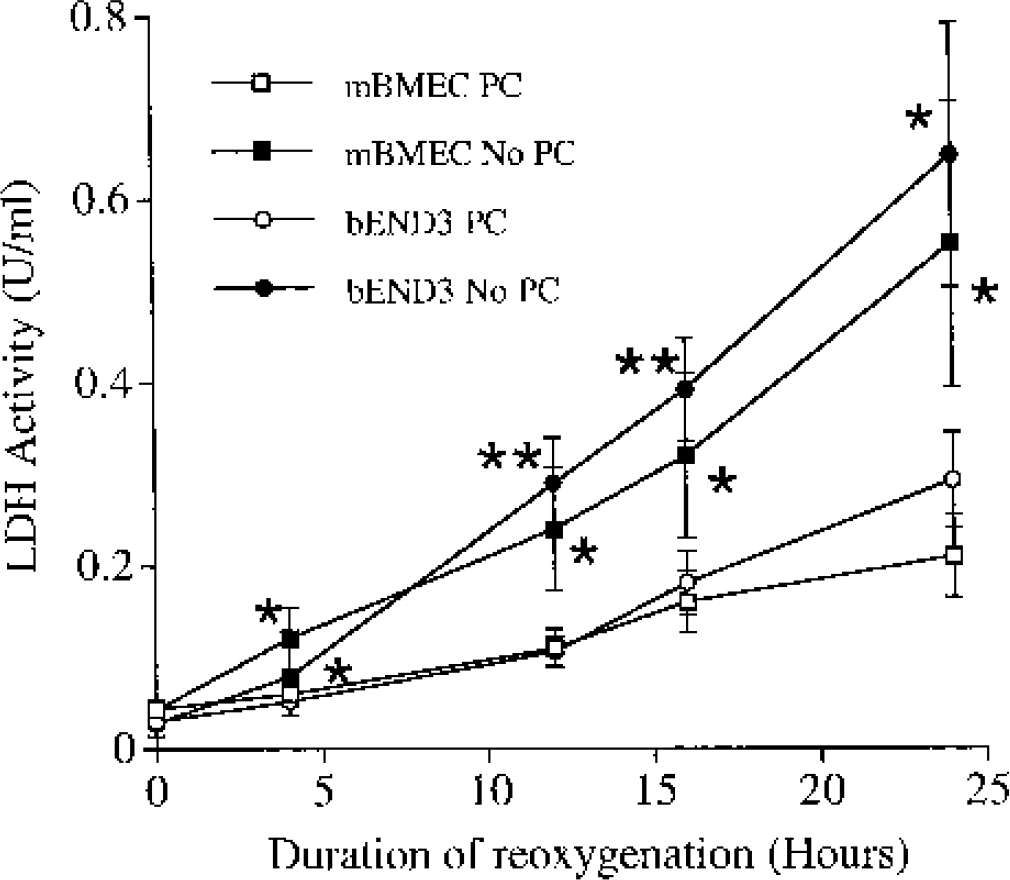
Effect of PC on media LDH activity in bEND.3 cells and mBMEC exposed to 5 hours of OGD and 24 hours of reoxygenation. bEND.3 cells and mBMEC were exposed to 1 hour of OGD (the PC stimuli) 1 day before exposure to 5 hours of OGD. The medium was replaced after the OGD and sampled for LDH activity measurements after 4,12, 16, and 24 hours of reoxygenation. Results were compared with cells of each type that had not been preconditioned (No PC). Values are mean ± SD; n = 3 separate cultures. Asterisks indicate significant differences from the corresponding No PC group at the ∗ P < 0.05 and ∗∗ P < 0.01 (two-tailed) levels.
Reoxygenation after 5 hours of OGD results in an upregulation in ICAM-1 expression in bEND.3 cells at both the mRNA and the protein level (Fig. 7 and Fig. 8). Preconditioning reduced both mRNA (Fig. 7) and protein (Fig. 8) upregulation. The repression of ICAM-1induction increased as the duration of the PC stimulus was lengthened from 15 to 60 minutes.
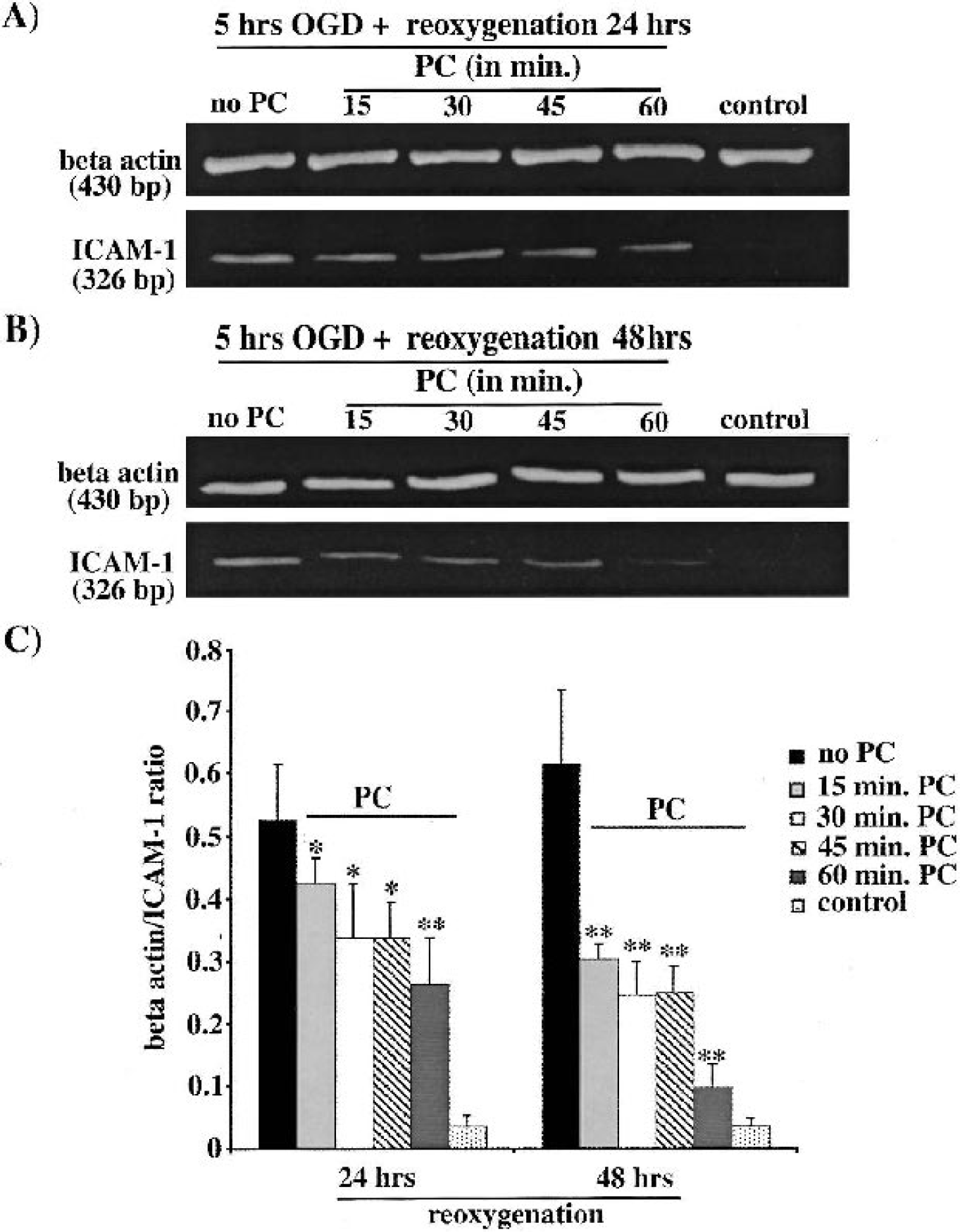
Example of a polymerase chain reaction analysis of ICAM-1 expression in bEND.3 cells exposed to 5 hours of OGD and either (
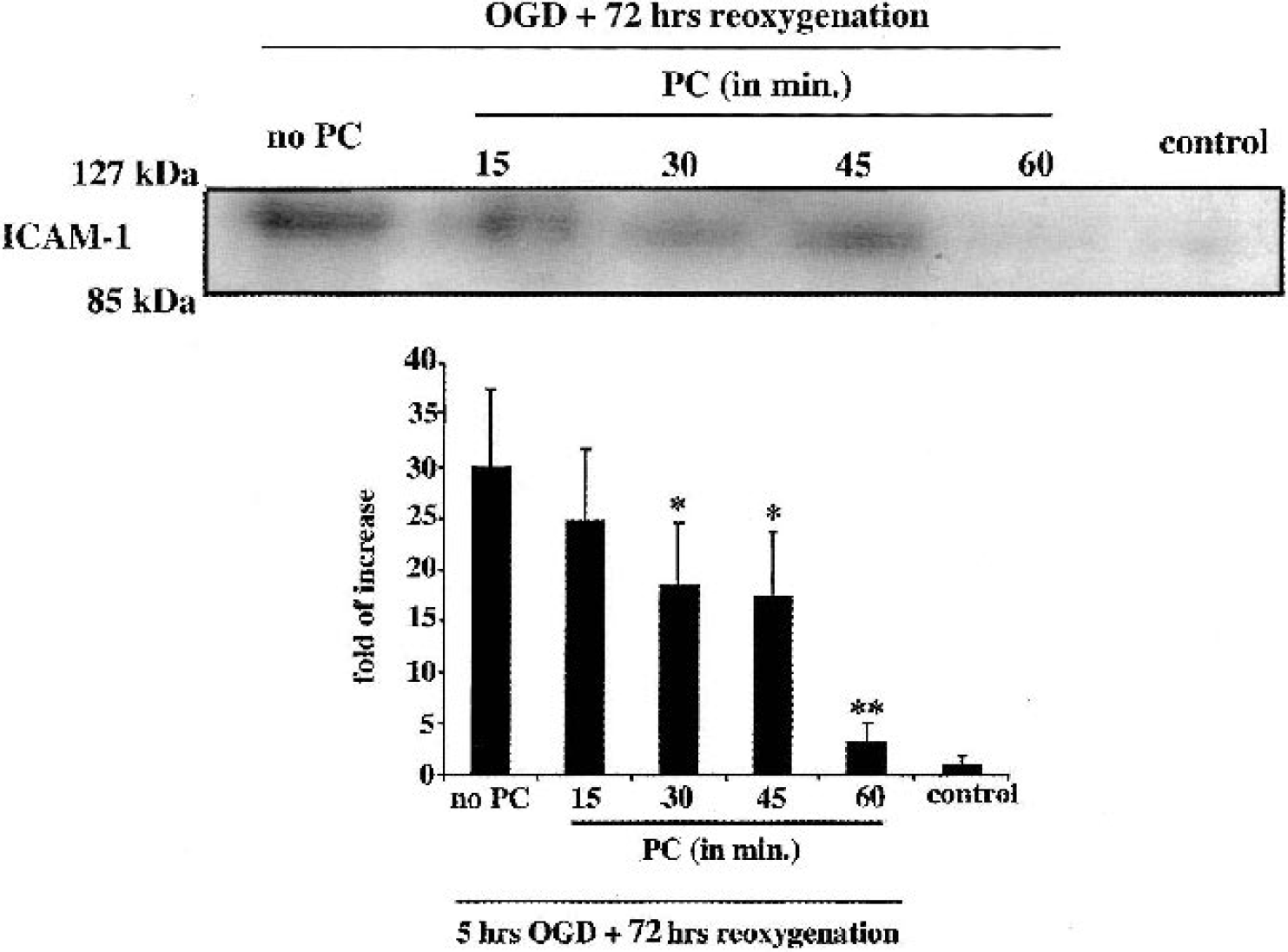
(
DISCUSSION
The current study indicates that PC, with a short duration of OGD, is capable of reducing the endothelial injury that results from a subsequent prolonged period of OGD and reoxygenation. In addition, PC reduced OGD-induced ICAM-1 upregulation. This suggests that with in vivo ischemia/reperfusion, ischemic PC may alter the inflammatory response by affecting the cerebral endothelium. In total, these results show that cerebral endothelial cells can be preconditioned in the absence of other cell types and suggest that this phenomenon may play a role in the overall effects of ischemic PC on the brain.
The current study used two different forms of cerebral endothelial cells, primary cultures of mBMEC and an immortalized mouse brain endothelial cell line. Currently, no cerebral endothelial cell culture system totally mimics the properties of cerebral endothelial cells in vivo. For example, in vivo, cerebral endothelial cells form the blood–brain barrier with a very high transendothelial electrical resistance, something that is not matched by even primary cultures in vitro (de Boer et al., 1999; Pardridge, 1998). Co-culturing brain endothelial cells with astrocytes results in a higher transendothelial electrical resistance, although still less than what is found in vivo (de Boer et al., 1999; Pardridge, 1998). However, for the current study, such co-cultures were not chosen because the presence of astrocytes might confound data interpretation; i.e., it would be uncertain whether any endothelial effect was direct or via astrocyte protection. Because of the low transendothelial electrical resistance of cerebral endothelial cell cultures, three other quantitative endpoints were chosen to determine whether PC was protective: LDH release, cell viability (using calcein-AM and EthD-1), and ICAM-1 upregulation. LDH release is a common marker of cell viability (Kaku et al., 1993; Kapinya et al., 2002; Perez-Pinzon et al., 1995), and the effects of PC on LDH were matched by morphologic observations on the endothelial cells (Fig. 1) and the cell viability assay (Fig. 4). ICAM-1 was chosen because endothelial ICAM-1 upregulation is an important part of the inflammatory response during ischemia/reperfusion (Berti et al., 2002; Feuerstein et al., 1997) and substantial evidence indicates that inflammation is an important part of secondary injury after stroke (Feuerstein et al., 1997; Hallenbeck, 1996). Examination of LDH release, cell viability, ICAM-1 upregulation, and morphologic changes all indicated that PC protects endothelial monolayers from injury during OGD with or without reoxygenation.
In terms of whether the in vitro results presented here reflect what would occur in vivo, it is important to note that the impetus for these experiments was our previous results indicating that ischemic PC protects the cerebral endothelium in vivo (Masada et al., 2001). Thus, qualitatively, there is agreement between in vitro and in vivo results. It should also be noted that two different types of cerebral endothelial cells (primary cultures and an immortalized cell line) gave almost identical results, although (as discussed previously) both models can be criticized as not fully reflecting the properties of cerebral endothelial cells in vivo. No astrocytes were present and the models used were static without the presence of hemodynamic forces that might alter endothelial function (Grant et al, 2002).
These experiments used OGD for both the PC stimulus and the injury inducing insult. For in vitro experiments, OGD is the closest approximation to the conditions that occur during cerebral ischemia, and OGD has been used in numerous “ischemia”-related studies (Kaku et al., 1993; Kapinya et al., 2002; Perez-Pinzon et al., 1995). However, there are numerous differences between ischemia and OGD (e.g., flow, changes in other metabolites, absolute changes in oxygen and glucose concentration). Although we have shown that ischemic PC protects the cerebral endothelium in vivo (Masada et al., 2001) and PC with OGD protects cerebral endothelial cells in vitro (this study), there may be a quantitative difference. In the current study, we found evidence of protection with 15 minutes of OGD as the PC stimulus, but the greatest protection was found with a 45 to 60 minutes of OGD (longer periods were not used because they cause endothelial damage). In vivo, we used 15 minutes of transient middle cerebral artery occlusion as the PC stimulus (Masada et al., 2001), but longer durations cause infarction.
The time course for PC-induced endothelial protection (Fig. 2) is similar to that of a number of studies on PC-induced reductions in brain infarct volume. In contrast to heart, where there is early and late protection (Carroll and Yellon, 1998; Rubino and Yellon, 2000), most but not all studies in brain have only shown late protection with PC (Barone et al., 1998; Chen et al., 1996; Perez-Pinzon, 2000); i.e., there has to be a time interval of about 24 hours between the PC stimulus and the injury inducing insult for there to be protection. This delay is because of the need for new protein synthesis (Barone et al., 1998), and it appears that this is also the case for protection of the cerebral endothelium.
Endothelial or vascular PC has received more attention in heart, although even there it is “an overlooked phenomenon” (Rubino and Yellon, 2000). In the coronary circulation, ischemic PC protects against ischemia/reperfusion-induced endothelial structural damage and loss of endothelium-dependent vasodilation (Kaeffer et al., 1996; Richard et al., 1994; Thourani et al., 1999). In heart and aortic endothelial cells, ischemic PC also reduces neutrophil infiltration after ischemia-reperfusion injury (Beauchamp et al., 1999; Kurzelewski et al., 1999; Thourani et al., 1999). This effect of PC appears to reflect changes in adhesion molecules. Thus, in aortic endothelial cells, ischemic PC prevents the upregulation of ICAM-1 (Beauchamp et al., 1999), whereas in human umbilical vein endothelial cells, PC markedly reduces E-selectin upregulation after exposure to tumor necrosis factor-α exposure (Zahler et al., 2000). The results from the current study indicate that PC has similar antiinflammatory effects on cerebral endothelial cells.
The cellular mechanisms involved in cerebral endothelial PC are yet to be investigated. Some information is available on coronary endothelium, although it should be noted before discussion that there may be difficulties in extrapolating between heart and brain because the cerebral endothelium is specialized, forming the blood–brain barrier. It should also be noted that there is early and delayed PC in coronary endothelium (Zhou et al 1996). In heart, there is evidence for a role for KATP channels (Bouchard and Lamontagne, 1996), adenosine (Giannella et al., 1997; Maczewski and Beresewicz, 1998), and bradykinin (Bouchard et al., 1998; Giannella et al., 1997) in triggering endothelial PC. Interestingly, there appear to be differences in the adenosine receptor subtypes involved in PC in endothelial cells (A2) and cardiomyocytes (A1 and A3) (de Jong et al., 2000). This also seems to be the case for bradykinin (Rubino and Yellon, 2000) and the signaling cascades involved in PC also appear to differ between cell types (Cepinskas et al., 2001). Should there be such cell-specific PC events in brain, this would provide a tool for examining the importance of endothelial PC in the overall protective effects of IPC on the brain.
Protecting the cerebral endothelium may affect overall ischemic brain damage in a number of ways. Thus, inhibiting blood–brain barrier disruption may reduce edema formation and hemorrhagic transformation on reperfusion. Preventing endothelial expression of adhesion molecules will affect postischemic inflammation. Reducing endothelial damage may also prevent alterations in the release of vasoactive substances by the endothelium that may participate in postischemic hypoperfusion (Bari et al., 1998; Takagi et al., 1977).
In conclusion, cerebral endothelial cells can be protected by preconditioning in vitro. This may play an important role in the overall effects of ischemic preconditioning in vivo (e.g., by reducing the influx of leukocytes). Understanding the mechanisms involved and how other cell types (e.g., astrocytes) influence the PC response may provide new insight into therapeutic strategies to limit cerebrovascular damage during ischemia.