
Abstract
Hyperoxia causes a transient decrease in CBF, followed by a later rise. The mediators of these effects are not known. We used mice lacking endothelial or neuronal nitric oxide synthase (NOS) isoforms (eNOS−/− and nNOS−/− mice) to study the roles of the NOS isoforms in mediating changes in cerebral vascular tone in response to hyperoxia. Resting regional cerebral blood flow (rCBF) did not differ between wild type (WT), eNOS−/− mice, and nNOS−/− mice. eNOS−/− mice showed decreased cerebrovascular reactivities to NG-nitro-L-arginine methyl ester (L-NAME), PAPA NONOate, acetylcholine (Ach), and SOD1. In response to hyperbaric oxygen (HBO2) at 5 ATA, WT and nNOS−/− mice showed decreases in rCBF over 30 minutes, but eNOS−/− mice did not. After 60 minutes HBO2, rCBF increased more in WT mice than in eNOS−/− or nNOS−/− mice. Brain NO-metabolites (NOx) decreased in WT and eNOS−/− mice within 30 minutes of HBO2, but after 45 minutes, NOx rose above control levels, whereas they did not change in nNOS−/− mice. Brain 3NT increased during HBO2 in WT and eNOS−/− but did not change in nNOS−/− mice. These results suggest that modulation of eNOS-derived NO by HBO2 is responsible for the early vasoconstriction responses, whereas late HBO2-induced vasodilation depends upon both eNOS and nNOS.
Elevated inspired concentrations of oxygen elicit cerebral vasoconstriction and decrease CBF in humans and other mammalian species (Bergo and Tysseboth, 1992; Omae et al., 1998; Torbati et al., 1978; Visser et al., 1996). Hyperoxic vasoconstriction may protect the brain from excess tissue oxygenation that causes central nervous system toxicity. Although this effect is well-recognized, its mechanism is not well-understood. Systemic inhibition of endothelial and neuronal nitric oxide synthases (eNOS and nNOS) by NG-nitro-L-arginine methyl ester (L-NAME) protects against oxygen toxicity (Oury et al., 1992) and attenuates cerebral vasoconstriction in rats exposed to hyperbaric oxygen (Demchenko et al., 2000b). These observations led us to speculate that NO is involved in HBO2-induced vasoconstriction because it has a constitutive vasorelaxing effect and participates in maintaining basal tone of cerebral vessels (Iadecola et al., 1994). Interference with the effect of NO on basal tone, for example, by NOS inhibition, can elicit vasoconstriction and lead to reductions in CBF. To evaluate this premise, we measured extracellular NO levels in rat striatum and found initial decreases in its interstitial concentration in relation to the vasoconstrictor response to HBO2 (Demchenko et al., 2000a). However, the relative contributions of NO derived from eNOS or nNOS isoforms to CBF responses to hyperoxia have been difficult to establish because NOS inhibitors lack adequate selectivity between isoforms. Moreover, at least four sites of NO production may contribute to the fall in NO levels during HBO2-induced cerebral vasoconstriction: endothelium, vascular nerves, parenchymal neurons, and glial cells. The development of knockout mice that lack eNOS or nNOS isoforms (eNOS−/− and nNOS−/− mice) provides an opportunity to evaluate independently the role of these isoforms in CBF responses to hyperoxia.
In this study, we performed physiologic experiments to evaluate and compare the effects of NO derived from endothelial and neuronal NOS on basal vascular tone and hyperoxic cerebral vasoconstriction. We measured rCBF responses to L-NAME, acetylcholine (ACh), PAPA NONOate and SOD1. We hypothesized that eNOS−/− mice would show less pronounced rCBF response to hyperoxia than nNOS−/− mice because hyperoxia promotes vasoconstriction by enhancing endogenous superoxide anion (O2−) generation and decreasing basal vasodilator effects of endothelial NO (Demchenko et al., 2002). We measured temporal profiles of the NO metabolites nitrite and nitrate (NOx) by intracerebral microdialysis and 3-nitrotyrosine (3NT) as a biochemical marker of peroxynitrite (ONOO−) formation in wild-type and NOS mutant mouse strains under conditions of hyperoxia.
MATERIALS AND METHODS
NOS−/− mutant mice
The mutant mouse strains used in this study were generated and described in detail previously (Huang et al., 1993, 1995). eNOS−/− mice were generated by homologous recombination and replacement of a gene segment encoding the NADPH ribose and adenine binding sites by a neomycin resistance gene. nNOS−/− mice were generated by replacement of the first two exons of nNOS. Both strains of knockout mice are viable, fertile, and have anatomically normal neuronal and vascular development. Both eNOS−/− and nNOS−/− mice were backcrossed to the C57BL/6 strain for more than ten generations to prevent genetic background effects. C57BL/6 WT mice were used as control animals.
Chemicals
All chemicals were obtained from Sigma Chemicals (St. Louis, MO, U.S.A.) unless otherwise indicated. PAPA NONOate [1-propamine, 3-(2 hydroxy-2-nitroso-1-propylhydrazine)] was purchased from Cayman Chemicals (Ann Arbor, MI, U.S.A.), and pancuronium bromide was obtained from Elkins-Sinn, Inc. (Cherry Hill, NJ, U.S.A.).
Surgical preparation
WT, eNOS−/−, and nNOS−/− mice (10–12 weeks of age) were housed under diurnal lighting conditions and allowed food and water ad libitum. The Institutional Animal Care and Use Committee at Duke University approved the surgical and anesthetic procedures. Mice were initially anesthetized with urethane (800 mg/kg, i.p.) followed by continuous i.p. infusion of one fourth of the initial dose per hour using a syringe pump (Model 351, Sage Instruments, Cambridge, MA, U.S.A.). The femoral artery and vein were catheterized for monitoring blood pressure, removing blood samples, and pharmacologic infusions. After ensuring appropriate anesthesia, a tracheotomy was performed, and mice were placed on a ventilator with 30% O2 (balance N2) and paralyzed with pancuronium bromide (0.5 mg/kg, i.v.). Each animal's head was fixed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA, U.S.A.), and hydrogen sensitive electrodes were inserted through a burr hole into the caudate putamen (CP) and parietal cortex (PC). Electrodes were made of platinum wire (100 μm diameter) insulated with epoxy, except for 1.0-mm length at the 15- to 20-μm pointed tip that was coated with a hydrophobic gas permeable Nafion membrane to prevent fouling. In some mice, a microdialysis probe (CMA/10: 2-mm membrane length, 0.24 mm O.D., CMA/ AB, Sweden) was placed into the CP and perfused continuously with artificial cerebrospinal fluid (CSF) using a CMA/100 microinjection pump. Body temperature was monitored with a thermal probe inserted into the rectum and maintained between 36°C and 37°C by a heating pad.
Quantitative measurements of regional CBF
Regional CBF in mice was measured by the hydrogen (H2) clearance method modified for hyperbaric conditions (Demchenko et al., 1998). Platinum electrodes inserted into the brain regions and reference Ag-AgCl electrodes at the base of the tail were used to generate H2 clearance curves for rCBF calculations. For this, H2 (2.5% in air) was added via the respirator for 60 seconds, after which gas containing H2 was switched to the base breathing gas, and H2-washout curves were recorded. Absolute values of rCBF (mL/100g/min) were calculated by the initial slope method using WINDAQ (D-1200 AC, DATAQ Instruments, OH, U.S.A.) and Mathematica 3.0 software (Wolfrom Research, Inc.) with minor modifications (Demchenko et al., 1998).
Measurement of interstitial NO availability
Extracellular NO from the yield of nitrite and nitrate (NOx) was estimated from microdialysis measurements conducted under hyperoxic conditions (Demchenko et al., 2000a). A microdialysis probe was perfused continuously in the CP at a flow rate 1.5 μL/min with artificial CSF using a CMA/100 microinjection pump (Carnegie Medicine AB, Stockholm, Sweden). After a 2-hour equilibration period, dialysate samples were collected with a CMA/140 microfraction collector every 15 minutes before and during exposure to HBO2.
After hyperbaric oxygen (HBO2) exposure, samples in volume of 20 μL were assayed for NOx using a catalytic method to reduce NO oxidation products to NO gas, which was then measured by chemiluminescence (NO Analyzer, Cervitex, MA, U.S.A.). Measurements of known concentration of nitrite and nitrate were quantitatively linear between 25 and 500 pmol/20 μL and were used to calibrate the samples. The fractional recovery of these two species was nearly identical; the recovery efficiency for the dialysis probes were 32 ± 6% and 31 ± 7% for NO2 and NO3, respectively. The data were expressed in μM concentrations of nitrite and nitrate (NOx).
Measurement of interstitial 3NT
In the brain, tyrosine residues react with peroxynitrite to yield 3NT, a stable byproduct that can be used as a marker of ONOO− formation (Beckman and Koppenol, 1996; Ischiropoulos et al., 1992). For detection of 3NT in the CP of mice exposed to HBO2, interstitial samples were collected every 15 minutes from microdialysis probes perfused continuously with artificial CSF at a flow rate 1.5 μL/min. After hyperbaric exposure, samples in volumes of 20 μL were assayed for 3NT content by HPLC on a setup containing a Shimadzu solvent delivery system (ESA model 580) and a reversed phase C-18 column (5-μm bead size; 4.6 × 150 mm; MCM). Isocratic elution was performed using a mobile phase of 20 mM sodium phosphate, 5 mM dodecyltriethylammonium chloride, and 10% methanol (pH 7.5). A dual-channel electrochemical detector (ESA, Inc., Coulochem II, Chelmsford, MA, U.S.A.) was set to +600 mV for channel A to quantify tyrosine and aminotyrosine as well as remove compounds that oxidize at lower potentials than 3NT (Skinner et al., 1997). Channel B was placed in series after channel A was set to +800 mV to detect 3NT. This optimizes detection of 3NT in samples with large amounts of tyrosine relative to 3NT. The retention time for 3NT was 7 minutes longer than for tyrosine, which allowed a further 1,000-fold enhancement of sensitivity to 3NT by programming a variable gain into the detector. A guard cell placed between the solvent delivery system and injector and set to +850 mV removed electrochemically active compounds from the mobile phase. 3NT was confirmed by retention time using authentic standards and by addition of 10 mM sodium hydrosulfite (dithionite) to 3NT, resulting in reduction to aminotyrosine and changes in retention time and electrochemical characteristics. The recovery efficiency for the dialysis probes were 29 ± 6% and 34 ± 7% for 3NT and tyrosine, respectively. The data were expressed as ratios of 3NT to tyrosine to normalize for different interstitial concentrations of tyrosine.
HBO2 exposure
Mice were instrumented with electrodes in a hyperbaric chamber (volume = 36 m3) containing the stereotaxic frame, respirator, pressure transducer, heating pad, infusion pump, and intracerebral microdialysis set-up. The electrical signals passed through the hull to amplifiers located outside the chamber. After 2 hours, once the mice were in a stable anesthetized state, three control H2 clearance curves were recorded, and three control dialysate samples were collected. The respirator circuit was then connected to a supply of 100% O2 and the chamber compressed with air to a total pressure of 5 ATA at a rate of 0.6 ATA per minute. The HBO2 exposures lasted 60 minutes, during which rCBF was measured every 15 minutes using a special, remotely operated pneumatic system for O2 and H2 delivery. MABP and body temperature were monitored continuously. Samples of dialysate were collected automatically every 15 minutes and used to measure NOx or 3NT after decompression. For constant anesthesia in the chamber, urethane (200 mg/kg per hour) was administered continuously by infusion pump and pancuronium bromide (0.1 mg/kg per hour) was given to prevent respiratory motion. Arterial pO2, pCO2, and pH were determined (IL model 1306 Blood gas/ pH analyzer, Lexington, MA, U.S.A.) before compression and immediately after decompression while the mice breathed 100% O2 at 1 ATA.
Experimental design
Three series of experiments were conducted. In series I, we tested the NO-dependent cerebrovascular reactivity in WT and NOS-mutant mice breathing 30% O2 (balance N2) at atmospheric pressure. Baseline rCBF was measured in WT (n = 9), nNOS−/− (n = 8), and eNOS−/− (n = 9) mice. rCBF was then measured in response to infusions of the NO donor PAPA NONOate (n = 9 each strain), ACh (n = 8 each strain), CuZn superoxide dismutase (SOD1, n = 9 each strain), and L-NAME (n = 8 each strain). In series II, time-dependent measurements of blood flow in CP and PC, as well as extracellular NOx accumulation in CP, were made in WT (n = 9), nNOS−/− (n = 9), and eNOS−/− (n = 8) mice during 60 minutes of HBO2 exposure at 5 ATA. Independently, in series III, interstitial 3NT levels in CP were measured in WT (n = 7), nNOS−/− (n = 5), and eNOS−/− (n = 6) mice during HBO2 exposure at 5 ATA. The purpose of series II and III experiments was to evaluate the relative contributions of eNOS and nNOS to rCBF responses to elevated brain pO2 and to correlate the time course of striatal blood flow with NO levels and ONOO− formation.
Statistical analysis
All values from groups of animals were expressed as mean ± SD. Absolute and percent changes in rCBF and physiologic variables (MABP and arterial blood gas values) in response to PAPA NONate, ACh, SOD1, and L-NAME treatment were compared with baseline values before treatment by paired t-tests. Changes in rCBF, NOx, and ONOO− over time under HBO2 conditions were compared using repeated measures analysis of variance (ANOVA) followed by a post hoc comparison (Fisher's exact test). A value of P < 0.05 was accepted as significant.
RESULTS
Roles of eNOS and nNOS isoforms in cerebrovascular reactivity
All anesthetized, artificially ventilated mice reported in this study were maintained at physiologic body temperature (36° to 37°C). Arterial blood pressure and resting rCBF did not significantly differ between WT, nNOS−/−, and eNOS−/− animals. To assess the roles of nNOS and eNOS in regulating resting rCBF, blood flow was measured in CP and PC in mice treated with the nonselective NOS inhibitor, L-NAME (30 mg/kg, i.p.) to suppress both endothelial and neuronal NO production. After 30 minutes, rCBF was reduced significantly in WT and nNOS−/− mice. eNOS−/− mice did not show statistically significant changes in PC perfusion and showed less change in CP perfusion than WT and nNOS−/− mice (Table 1). These results indicate that eNOS-derived NO normally plays an important role in rCBF regulation and that in its absence, alternative mechanisms compensate to maintain rCBF, but are not sensitive to L-NAME.
Blood pressure in wild type and NOS mutant mice treated with NO donor or acetylcholine
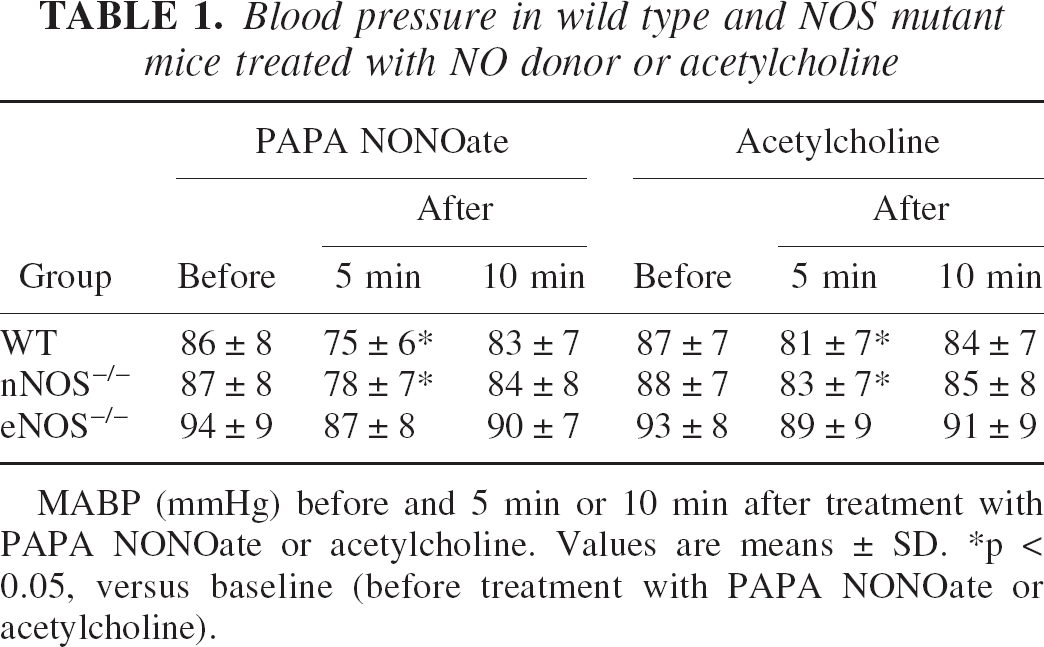
MABP (mmHg) before and 5 min or 10 min after treatment with PAPA NONOate or acetylcholine. Values are means ± SD.
p < 0.05, versus baseline (before treatment with PAPA NONOate or acetylcholine).
Because endothelium-derived relaxation is a physiologic response of blood vessels to stimuli that activate NO signaling, we next determined the extent to which nNOS or eNOS contribute to cerebrovascular reactivity in response to an NO donor (PAPA NONOate) and a physiologic stimulator of eNOS (ACh). PAPA NONOate (0.01 mg/kg, i.v.) increased rCBF in all three groups of mice, but the rCBF responses in eNOS−/− mutants were significantly less than in WT mice (Fig. 1A). ACh (0.05 mg/kg, i.v.) significantly augmented rCBF in WT and nNOS−/− mice but not in eNOS−/− mice (Fig. 1B). In mice given NO donor or ACh, mean arterial blood pressure decreased slightly and transiently by 10 to 15 mmHg for less than 5 minutes (Table 2). In both cases, rCBF measurements were made 10 minutes after NO donor or ACh administration when MABP had returned to pretreatment levels.
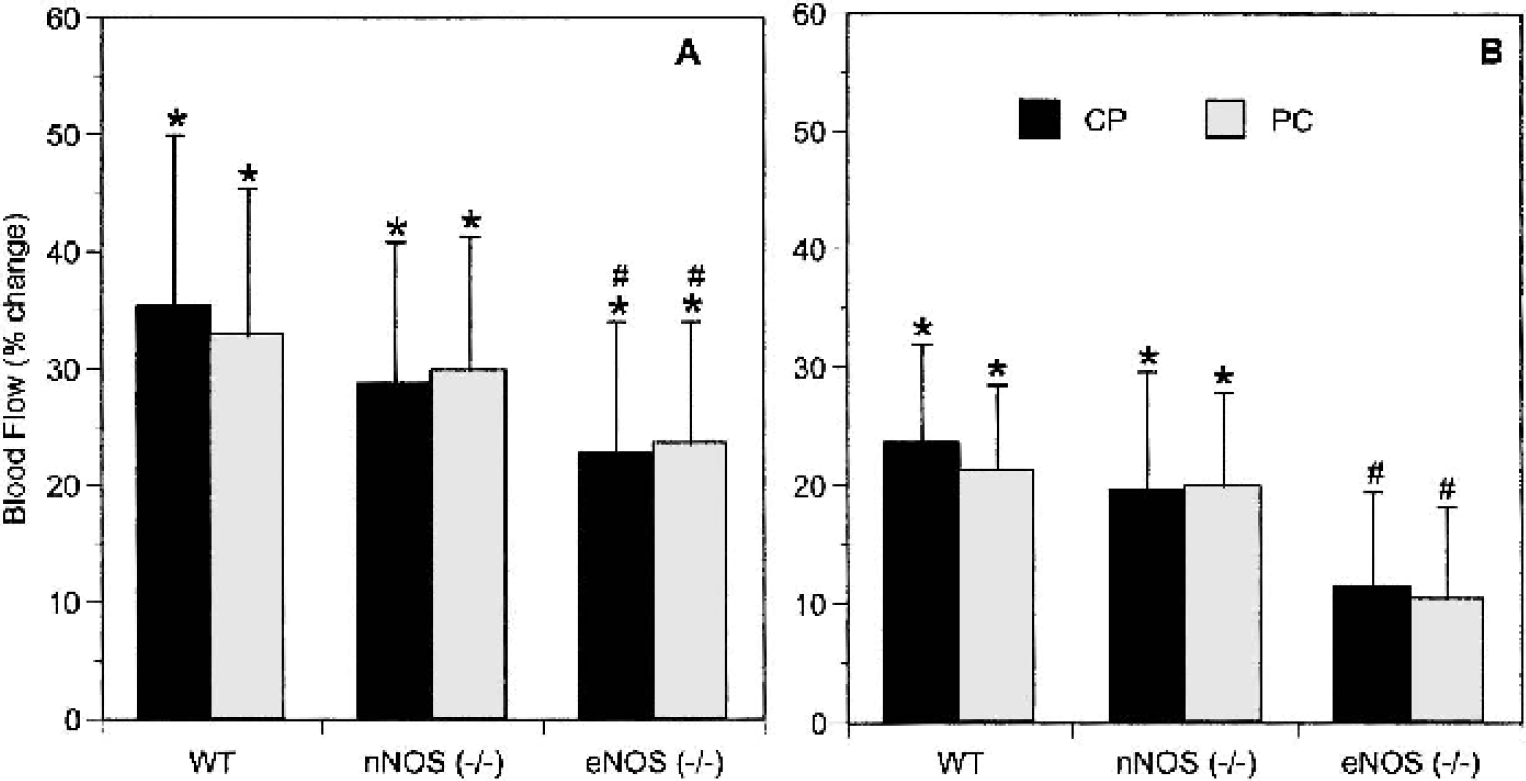
NO-dependent cerebrovascular responses in WT and NOS−/− mutant mice.
Arterial blood gases
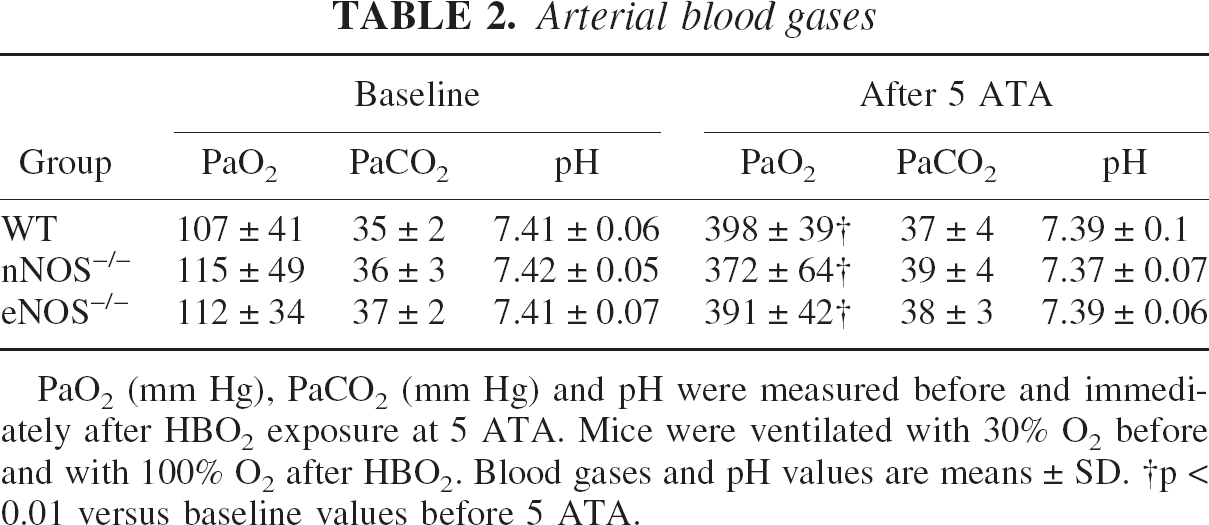
PaO2 (mm Hg), PaCO2 (mm Hg) and pH were measured before and immediately after HBO2 exposure at 5 ATA. Mice were ventilated with 30% O2 before and with 100% O2 after HBO2. Blood gases and pH values are means ± SD.
p < 0.01 versus baseline values before 5 ATA.
NO bioactivity in cerebral vessels under basal conditions depends in part upon its interaction with O2−, which reacts with and inactivates NO (Didion et al., 2001; Wei et al., 1985). We tested the effect of SOD1 infusion, which would reduce O2− at the surface of the vascular endothelium, upon rCBF. We previously showed that active, but not inactive, SOD1 reproducibly and significantly increases rCBF in rats after intravenous infusion (25 U/g body weight), reaching maximal amplitude within 10 minutes (Demchenko et al. 2000b). Using the equivalent dose and time parameters in mice, SOD1 significantly increased rCBF in the CP and PC in WT and nNOS−/− mice but not in eNOS−/− mice. The differences in CP rCBF responses between eNOS−/− mice and WT or nNOS−/− mice were statistically significant (Fig. 2). These results indicate that the eNOS isoform plays a key role in baseline regulation of vascular tone in cerebral blood vessels. By scavenging O2−, SOD1 increases NO bioactivity and CBF in WT and nNOS−/− mice (which express eNOS) but not in eNOS−/− mice (which lack eNOS).
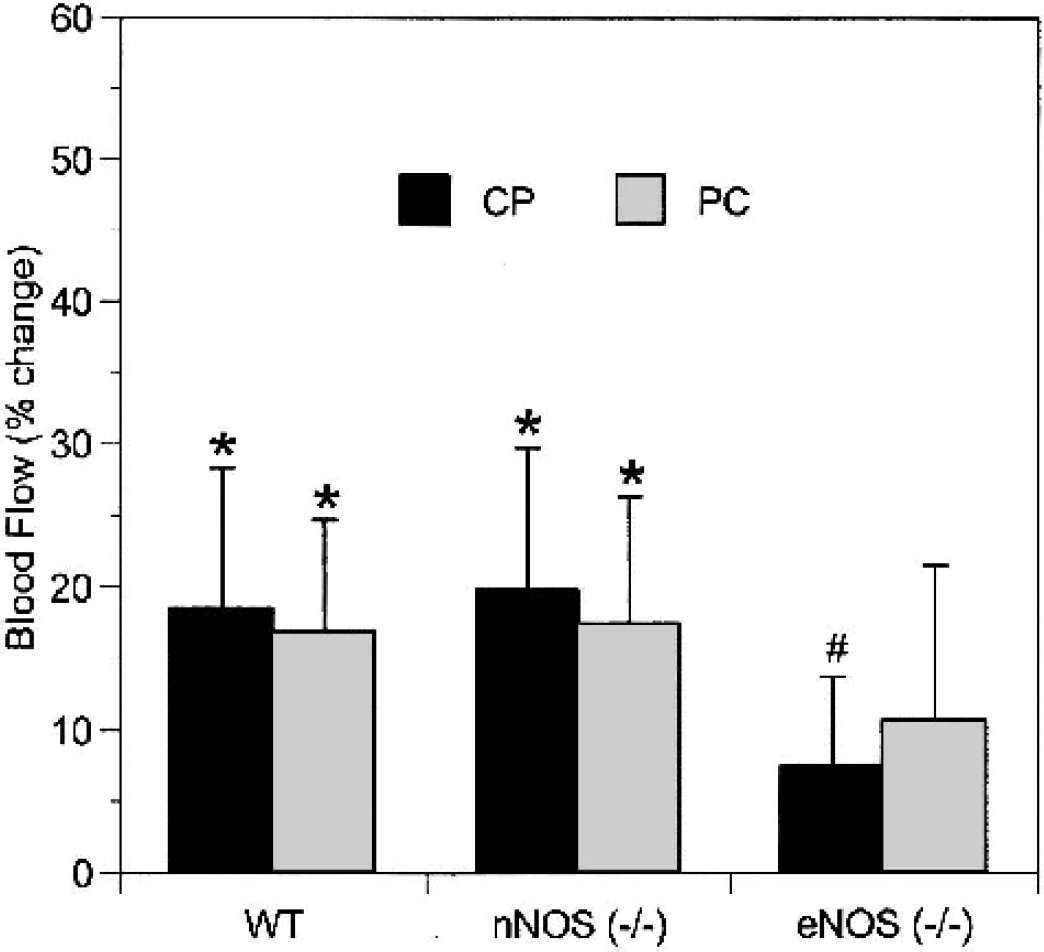
Cerebrovascular responses to SOD1 infusion. Blood flow in the CP and PC increased after intravenous SOD1 injection (25 U/g of body weight) in WT and nNOS−/− mice but not in eNOS−/− mice. Values are mean ± SD. ∗ P < 0.05 versus pretreatment; #P < 0.05 compared with WT or nNOS−/− mice.
Contribution of eNOS and nNOS in rCBF responses to HBO2
We assessed cerebrovascular responses to hyperoxia by exposing WT and NOS−/− mice to HBO2 at 5 ATA. In all three strains of animals, arterial pCO2 and pH values were in the physiologic range before and after 60 minutes of HBO2 (Table 3). It was not technically possible to measure blood gases and pH under HBO2, as the animals were in the chamber during this period. However, the high arterial pO2 values immediately after hyperbaric hyperoxia, with normal pCO2 and pH values, indicate that pulmonary gas exchange was preserved throughout the experiment and that high arterial pO2 values were attained during HBO2 exposure. MABP rose during chamber compression and for a few minutes of HBO2 exposure by 19% to 37% and remained elevated until decompression. HBO2-induced changes in MABP did not significantly differ between WT, nNOS−/−, and eNOS−/− mice. Postmortem examination did not reveal evidence for pulmonary hemorrhage or other tissue damage from the hyperoxic stress.
Blood pressure and rCBF response to L-NAME
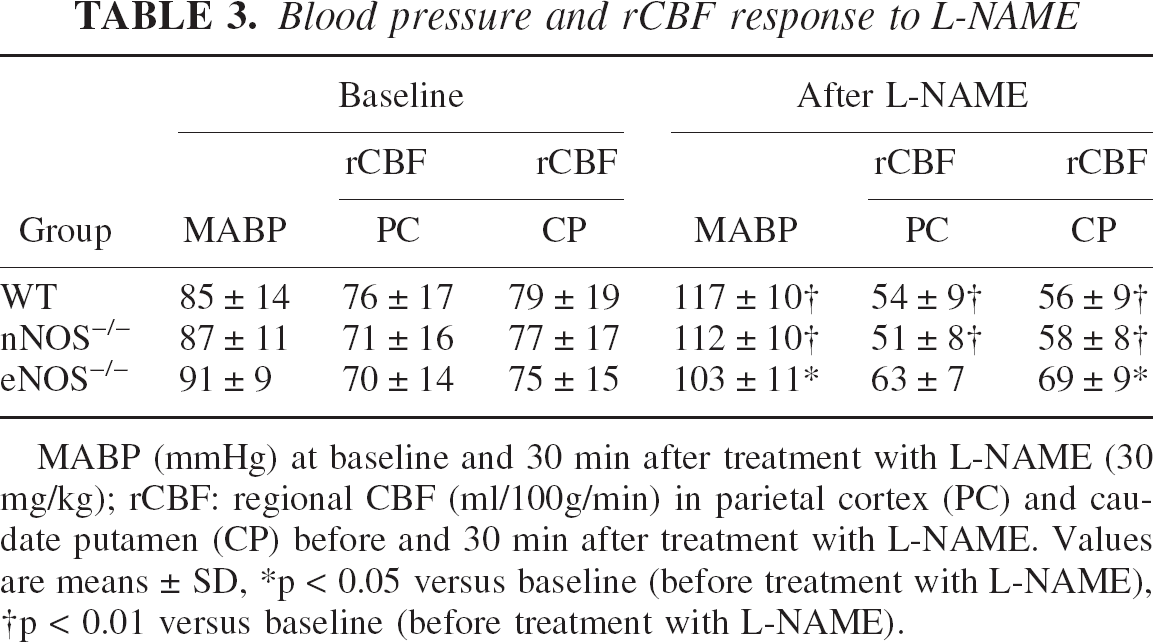
MABP (mmHg) at baseline and 30 min after treatment with L-NAME (30 mg/kg); rCBF: regional CBF (ml/100g/min) in parietal cortex (PC) and caudate putamen (CP) before and 30 min after treatment with L-NAME. Values are means ± SD
p < 0.05 versus baseline (before treatment with L-NAME)
p < 0.01 versus baseline (before treatment with L-NAME).
In WT mice, HBO2 at 5 ATA reduced rCBF over 30 minutes, but then blood flow rose gradually. By 45 minutes, rCBF had returned to preexposure levels, and then over the next 15 minutes, it increased above baseline levels (Fig. 3). Like WT mice, nNOS−/− mice showed significant cerebral vasoconstriction the first 30 minutes. In contrast, eNOS−/− mice did not show significant cerebral vasoconstriction. These results suggest that early HBO2-induced vasoconstriction is mediated by inhibition of basal vasodilation by eNOS (which is present in nNOS−/− and WT mice but is absent in eNOS−/− mice). Later, at 45 and 60 minutes, both nNOS−/− and eNOS−/− mice show hyperemia, though not as marked as seen in WT mice. These results suggest that the late hyperemia depends on both eNOS and nNOS, as it is reduced in mice lacking either isoform. These HBO2 effects were more pronounced in the striatum as compared with the cortex.
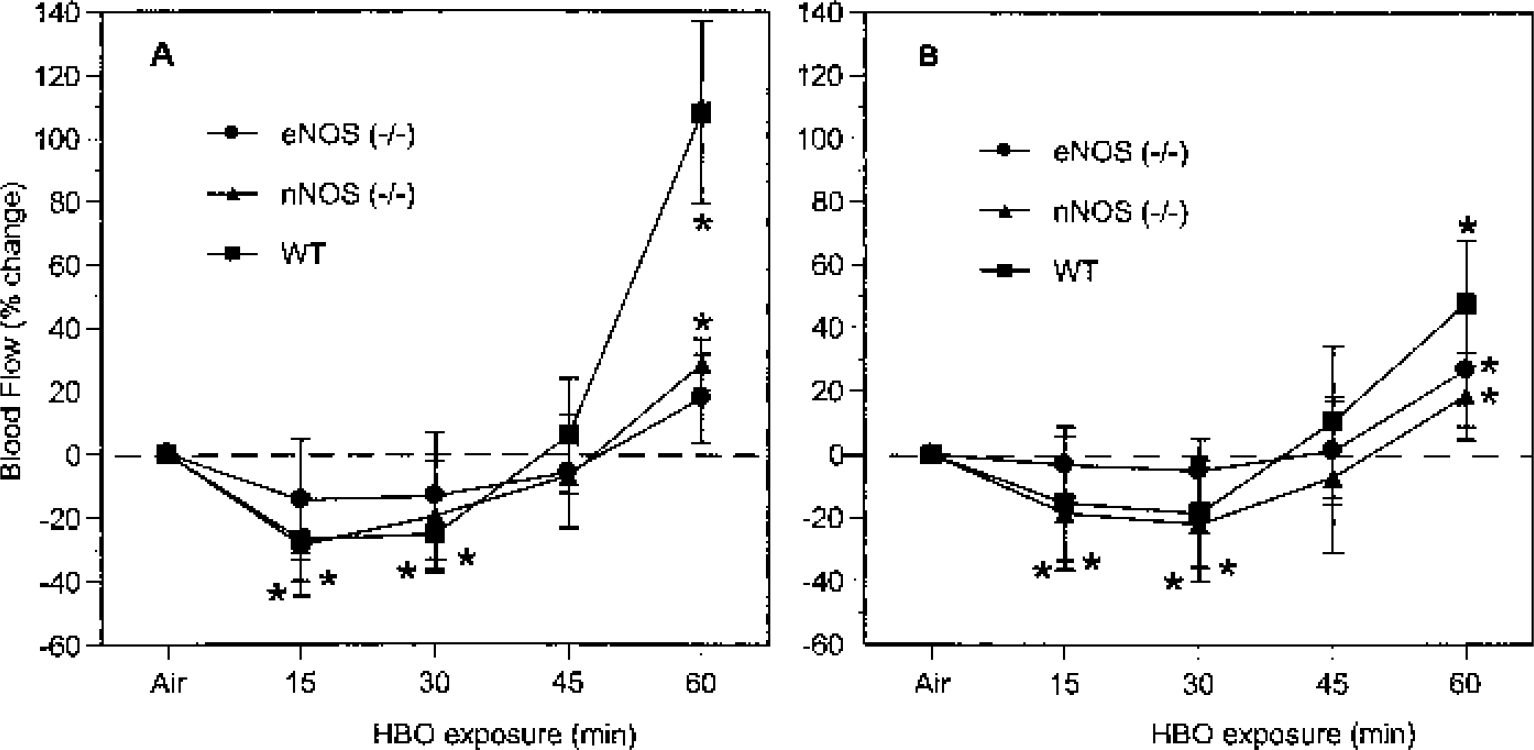
rCBF responses to HBO2. rCBF changes in CP
Changes in NOx and 3NT during HBO2 exposure
We used intracerebral microdialysis to measure interstitial NOx as a reflection of NO levels and 3NT to assess ONOO− formation in mice exposed to HBO2 at 5 ATA. The basal concentration of interstitial NOx in the striatum of WT mice was stable at 5.5 ± 1.6 μM. Within 30 minutes of HBO2 at 5 ATA, brain interstitial NOx decreased significantly, but thereafter it increased to above control levels (Fig. 4A). In nNOS−/− mice, the basal concentration of NOx was 3.9 ± 0.8 μM, which was significantly less than in WT strains (P < 0.05). Striatal NOx in nNOS−/− mice varied much less than in WT mice, suggesting that the measured changes in WT mice were caused in large part by changes in nNOS-derived NO. In eNOS−/− mice, NOx levels paralleled those in WT mice but were lower at all times, again consistent with nNOS as the source of NO in these changes.
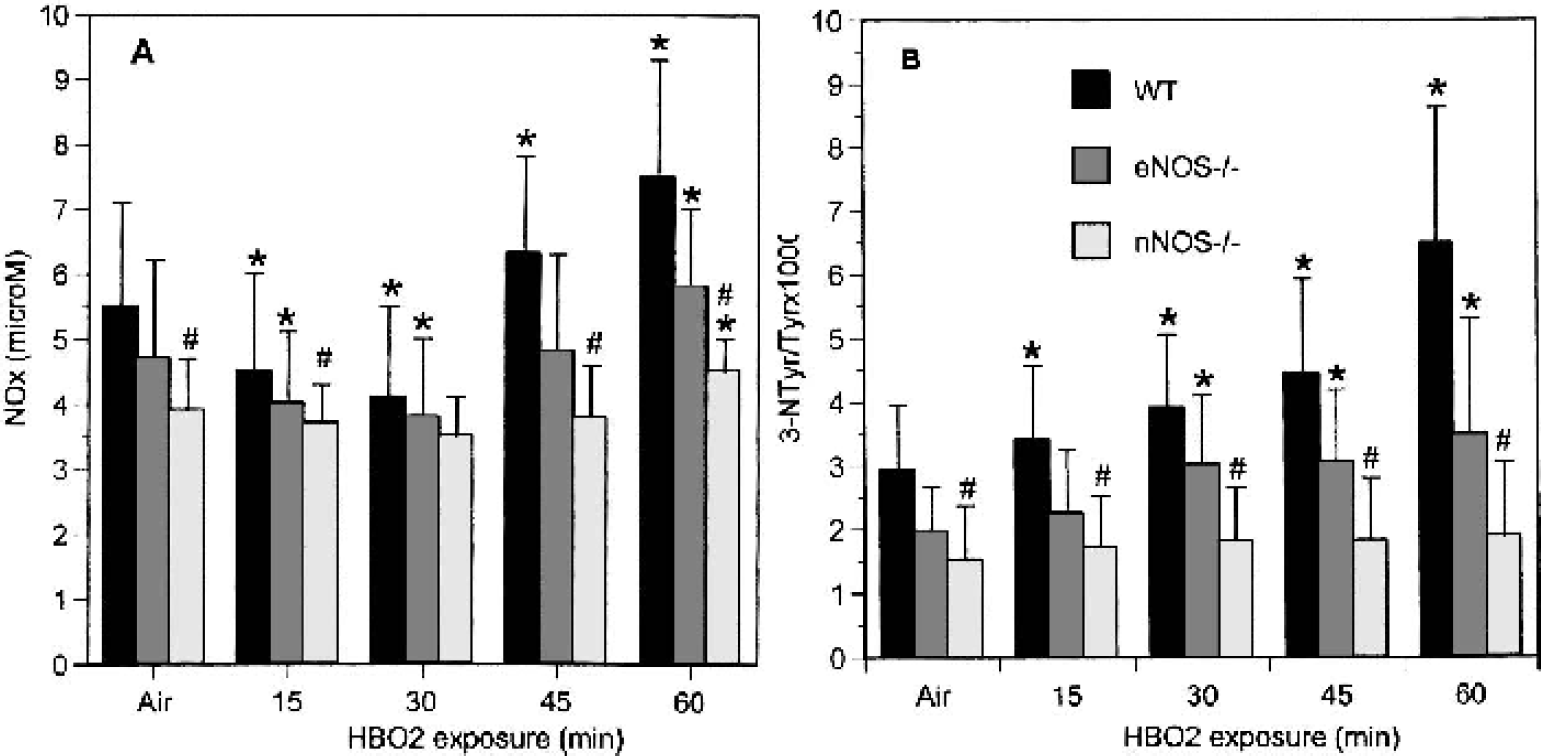
Effects of HBO2 on brain NOx and 3NT.
Brain interstitial 3NT content, unlike NOx, accumulated steadily during HBO2 at 5 ATA in WT and eNOS−/− mice (Fig. 4B). Marked increases in 3NT were detected during hyperbaric exposure in WT and eNOS−/− mice (both of which express nNOS) but not in nNOS−/− mice, which is consistent with nNOS being the predominant source of NO in the formation of ONOO−. The difference between 3NT levels in WT and eNOS−/− mice likely represents loss of vascular NO in the eNOS−/− mice. Indeed, nNOS−/− mice show a detectable and relatively constant level of 3NT, which represents the contribution of eNOS-derived NO to the total amount of OONO− formed. However, it is important to note that 3NT is an indirect marker for the presence of peroxynitrite, and it is normalized to total tissue tyrosine.
Overall, these results indicate that the nNOS isoform accounts for the major source of NO in the brain and for the variation in NOx levels and 3NT level. However, the blood flow results indicate that effects on eNOS-dependent blood flow mediate the early vasoconstriction seen in HBO2.
DISCUSSION
The present study of genetic knockout mice provides strong evidence implicating endogenous NO production in the regulation of brain blood flow during hyperoxia. These in vivo observations show that NO derived from endothelial and neuronal NOS contribute differently to basal vascular tone and to rCBF responses to hyperoxia. The principle findings supporting this premise were obtained from quantitative rCBF measurements in WT, eNOS−/−, and nNOS−/− mice.
First, we studied the rCBF response of WT and NOS−/− mice to the NOS inhibitor L-NAME, the NO donor PAPA NONOate, the endothelium-dependent vasodilator ACh, and SOD1. L-NAME significantly decreased resting rCBF in WT and nNOS−/− mice breathing 30% O2 at normal barometric pressure. However, it did not change cortical blood flow, and it decreased striatal perfusion less in eNOS−/− mutants. These data are consistent with earlier observations suggesting that basal production of endothelial NO influences cerebral vascular tone (Ma et al., 1996). The fact that basal rCBF values in eNOS−/− mice are normal indicates the existence of compensatory pathways of CBF regulation that keep CBF parameters in the physiologic range after eNOS gene deletion.
eNOS−/− mice treated with PAPA NONOate also show less pronounced increases in rCBF than do WT or nNOS−/− animals. These results suggest that brain vessels in eNOS−/− mice have reduced responsiveness to NO. Chronic absence of eNOS may reduce NO sensitivity of cerebral vessels, due to changes in downstream effector mechanisms like guanylate cyclase. As expected, rCBF responses to ACh were greater in WT and nNOS−/− mice than in eNOS−/− mice. These results agree with earlier data that arteriolar dilation to ACh superfusion was greater in nNOS−/− mice as compared with eNOS−/− mice (Meng et al, 1996).
Intravenous SOD1 infusion increases rCBF in WT mice, confirming an effect previously observed in rats (Demchenko, 2000b). This effect is preserved in nNOS−/− mice but is markedly blunted in eNOS−/− mice, indicating that vasoactivity of SOD1 depends upon the presence of eNOS-derived NO. Conversely, these results also suggest that bioactivity of endothelial NO may be regulated by superoxide anion levels in the brain vasculature (Wei et al.,1985). Because eNOS−/− mice lack endothelial NO, enhanced scavenging of O2− by SOD1 does not augment relaxation (Demchenko et al, 2000).
The differences in cerebrovascular reactivity to the NO donor and SOD1 between eNOS−/−and nNOS−/− mice were small, so we are cautious in attributing biologic relevance to them. However, the eNOS−/− responses to these agents were significantly decreased compared with those of WT and nNOS−/− mice. These findings indirectly support previous reports that NO bound to hemoglobin via thiol groups may regulate vascular tone in a manner dependent upon blood pO2 (Stamler et al., 1996). Indeed, the fact that intravenous SOD1 does not cross the blood-brain barrier implies that its actions may take place outside brain tissue, for example, in the endothelium and cerebral circulation, where SOD may prevent scavenging of NO by O2−, allowing it to bind to hemoglobin. However, further work will be necessary to strengthen this observation and to test this hypothesis directly. Overall, the results of cerebrovascular reactivity testing indicate that eNOS−/− and nNOS−/−mice exhibit different rCBF responses to stimuli activating NO signaling and that modulation of NO derived from eNOS is involved in NO-dependent vasoconstriction responses.
The rCBF response to HBO2 at 5 ATA was time-dependent and had a biphasic profile in WT mice, which is in agreement with previous studies in rats (Chavko et al., 1998; Demchenko et al., 1998). Initial reductions in rCBF (vasoconstriction) were followed by later elevations of blood flow above baseline levels (late vasodilation). nNOS−/− mice had responses qualitatively similar to WT mice, whereas eNOS−/− mice did not show significant HBO2-induced vasoconstriction. These results demonstrate that eNOS-derived NO is essential for HBO2 induced vasoconstriction. As shown in Fig. 3, the eNOS−/− response in the striatum was greater than in the cortex, but neither response was statistically significant. Further experiments will be necessary to determine whether there is a true difference in HBO2 vasoconstriction between the CP and the PC and, if so, whether it is mediated by other vasodilators or nNOS.
NOx measurements in the rat striatum demonstrated transient decreases in NOx recovery by microdialysis in HBO2 conditions (Demchenko et al., 2001). The directional changes in NO metabolites match those of O2-dependent rCBF responses, so collectively, these measurements provide experimental support for HBO2 vasoconstriction in the brain caused by interference with local NO levels in vivo. However, the enzymatic source of NO was not known nor was it clear that NOx measurements made by microdialysis measured vascular NO relevant to rCBF changes. Indeed, the results presented here suggest a discordance between the NOS isoforms responsible for brain NOx levels and rCBF changes. We found that WT and eNOS−/− mice show a transient decrease in interstitial NOx over 30 minutes of HBO2 exposure followed later by an increase in NOx to above control levels. In comparison, nNOS−/− mice consistently show minor and statistically insignificant changes in striatum NOx levels during 45 minutes of HBO2 exposure. Thus, nNOS is responsible for the HBO2 variation in NOx levels, whereas eNOS appears to be required for early HBO2 vasoconstriction.
Because NO is an endogenous vasodilator, there are theoretical reasons why reduced NO availability would lead to vasoconstriction and hence decreased CBF. Given our results on the effects of SOD1 on rCBF, a reasonable explanation for the transient decrease in NO is its inactivation by O2 (Demchenko et al., 2000a). In support of this, SOD1 reverses HBO2-mediated cerebral vasoconstriction in rats or mice (Demchenko et al., 2000a, 2002). NO can be scavenged by O2− to form ONOO− at a nearly diffusion-limited rate (Squadrito and Pryor, 1995). Increased rate of O2− generation by HBO2 can effectively reduce the availability of endothelium-derived NO. This predicts that NO bioactivity is regulated directly by O2−, rather than by independent and opposite effects of NO and O2− on vascular tone. The absence of O2−-dependent cerebral vasoconstriction in eNOS−/− mice supports this concept.
The initial decrease in interstitial NOx was followed by a subsequent increase to above control values in concert with increases in rCBF. This observation supports the concept that prolonged HBO2 exposure promotes NO production and augments rCBF (Demchenko et al., 2001). Because eNOS−/− and nNOS−/− mice show less late hyperemia after HBO2 than WT mice, late vasodilation appears to depend upon both endothelial- and neuronal-derived NO.
The results on NOx levels in WT, eNOS−/−, and nNOS−/− mice show the relative contribution of eNOS and nNOS isoforms in regulation of rCBF in HBO2 conditions. The absolute level of nNOS-derived NO in the brain far outweighs eNOS-derived NO. These data are consistent with our previous results (Hara et al., 1996). However, both endothelial (Ma et al., 1996) and neuronal (Gotoh et al., 2001) sources of NO are involved in maintenance of CBF. The nNOS inhibitor 7-nitroindazole has been reported to reduce resting CBF (Gotoh et al., 2001), presumably by inhibiting NO delivered from neurons and vascular nerves. However, eNOS-derived NO is important for regulation of CBF, and its modulation by HBO2 is responsible for the early vasoconstriction responses. The late marked increase of NOx in WT mice after 45 minutes of HBO2 exposure results from both nNOS- and eNOS-derived NO, since eNOS−/− and nNOS−/− animals have less NOx as compared with WT animals.
In conclusion, we present new evidence that HBO2 interferes with the constitutive relaxing effect of eNOS-derived NO on brain vessels in vivo. These cerebrovasoconstriction effects may be mediated by inactivation of endothelial NO by superoxide anion, leading to decreased CBF during HBO2 exposure. The late HBO2-induced vasodilation and hyperemia depend upon both eNOS- and nNOS-derived NO.