
Abstract
Atrial natriuretic peptide (ANP) plays an important role in the regulation of water and sodium in the body via cyclic GMP (cGMP) pathway. Although ANP has been shown to be protective in cerebral ischemia or intracerebral hemorrhage, its role in traumatic brain injury (TBI) has yet to be elucidated. We herein assessed ANP effects on brain water and sodium in TBI. Controlled cortical impact (3 mm depth, 6 m/sec) was used to induce an experimental cortical contusion in rats. Continuous administration of ANP 0.2 (n = 6) or 0.7 μg/kg/24 h (n = 6), cGMP analogue (8-Bromo-cGMP) 0.1 (n = 5) or 0.3 mg/kg/24 h (n = 5), or vehicle (n = 6) was begun 15 minutes after injury, using a mini-osmotic pump implanted into the peritoneal cavity. At 24 hours after injury, ANP significantly exacerbated brain edema in the injured hemisphere in a dose-dependent manner while it reduced brain sodium concentrations in both hemispheres. These ANP effects could be mimicked by a cGMP analogue. In the second series (n = 20), BBB integrity was assessed by evaluating the extravasation of Evans blue dye. ANP or cGMP analogue significantly worsened BBB disruption in the injured hemisphere at 24 hours after injury. These findings suggest that ANP administration exacerbates brain edema after the experimental cortical contusion in rats, possibly because of an increase in the BBB permeability via cGMP pathway, whereas it reduces brain sodium levels.
Keywords
Atrial natriuretic peptide (ANP), a member of a recently discovered natriuretic peptide family, is considered to play an important role in the regulation of water and sodium in the body by its abilities of natriuresis, diuresis, and vasorelaxation (Levin et al., 1998; Walther et al., 2002; Wilkins et al., 1997). It has recently been suggested that these actions require the binding of ANP with guanylyl cyclase receptors on the cell membrane, thus activating a cyclic GMP (cGMP) pathway (Light et al., 1989; Potter and Hunter, 2001). The activated cGMP subsequently stimulates cGMP-dependent protein kinases (PKG), which have a potential for inhibiting an amiloride-sensitive sodium absorption (Ibaragi et al., 1989; Sarker and Fraser, 2002). Thus ANP-induced natriuresis can be explained by the inhibition of tubular sodium reabsorption in the kidney (Jin et al., 2001; Light et al., 1989). ANP also modulates unidirectional transport of sodium from blood to brain via the cGMP-PKG pathway by prohibiting amiloride-sensitive Na+/H+ exchange (Ennis et al., 1996; Ibaragi et al., 1989). These findings suggest that ANP administration may reduce tissue sodium in brain injury.
Recent experimental studies in vitro showed that ANP increased the permeability of brain capillaries via the cGMP-PKG pathway (Grammas et al., 1991; Sarker and Fraser, 2002; Vigne and Frelin, 1992). It is therefore possible that ANP administration may exacerbate brain edema formation found in brain injuries, as based upon the vasogenic mechanism, and this may be enhanced by the preexistence of critical damage in the blood brain barrier (BBB). Recent evidence, however, showed that ANP administration not only decreased brain sodium levels but also attenuated brain edema in the rat models of cerebral ischemia (Nakao et al., 1990; Naruse et al., 1991) or intracerebral hemorrhage (Rosenberg and Estrada, 1995; Rosenberg et al., 1992). These effects on brain water may be attributed to the modulation of sodium transport in brain capillaries (Ennis et al., 1996; Ibaragi et al., 1989).
On the other hand, little is known about ANP effects on brain edema in traumatic brain injury (TBI). This is critical because brain edema is commonly believed to contribute to the high rate of secondary morbidity and mortality found in patients with TBI. We have recently shown that BBB opens after TBI, especially controlled cortical impact (CCI) (Beaumont et al., 2000), thus possibly indicating that ANP may influence brain water in TBI differently than in other models. Consequently, we herein assessed ANP effects on brain water and sodium in the experimental rat model of cortical contusion. To elucidate the mechanism, we examined whether an administration of cGMP analogue could mimic the ANP action and evaluated the BBB integrity by assessing extravasation of Evans blue dye. Mini-osmotic pumps implanted into the peritoneal cavity were used so that its content would be systemically delivered for 24 hours (Rosenberg and Estrada, 1995). To the best of our knowledge, this is the first report using an ANP administration in TBI.
MATERIALS AND METHODS
Experimental protocol and group
All surgical procedures were considered and approved by VCU Institutional Animal Care and Use Committee Regulations in compliance with NIH standards and guidelines. The present study included two different series as shown in Table 1. In the first series, 33 adult male Sprague-Dawley rats weighing 350 to 430 g were randomly divided into six groups (Table 1) to assess the effect of ANP or cGMP analogue on brain water and electrolytes. To evaluate the BBB integrity, an additional 20 rats were randomized into four groups (series 2, Table 1). Each surgery was performed without any knowledge of the group to which the animal belonged so as to maintain a blind study for analysis purposes.
Experimental groups
ANP, atrial natriuretic peptide; cGMP, cyclic GMP; cGMP analogue, 8-bromo-cGMP (membrane-permeable); BBB, blood brain barrier.
Surgical procedure and injury
All rats were given free access to food and water before surgery. General anesthesia was induced with 4% halothane. Animals were then intubated under direct vision and mechanically ventilated receiving a gas mixture of 70% nitrous oxide, 30% oxygen, and 1 to 1.5% halothane. Each animal was placed in a stereotaxic frame to secure the head, and a midline scalp incision was made so that a craniotomy could be performed to expose the right hemisphere of the brain. The CCI (3 mm depth, 6 m/sec) was used to induce an experimental cortical contusion as described previously (Beaumont et al., 2000; Dixon et al., 1991). The removed skull section was then replaced, followed by closure of the scalp.
Drug preparation and pump implantation
Continuous systemic administration of ANP, cGMP analogue, or vehicle was achieved via a mini-osmotic pump (model 2001D; ALZET Osmotic Pumps, DURECT Corporation, Cupertino, CA, U.S.A.). Briefly, a 1.5-cm midline skin and muscle incision was made on the abdomen. Fifteen minutes after injury, a pump filled with either drug or vehicle was implanted into the peritoneal cavity, followed by closure of the muscle and skin. The animal was then allowed to recover from the anesthesia.
This model of pump is designed to deliver its content at a uniform rate of 8.0 μL/h for 24 hours. The intraperitoneal administration of ANP with this pump was reported in the recent experimental study (Rosenberg and Estrada, 1995), in which ANP reduced brain water and sodium in the rat model of intracerebral hemorrhage. In accordance with that study (Rosenberg and Estrada, 1995), two different doses of ANP (Sigma Chemical Co., St Louis, MO, U.S.A.), 0.2 (n = 6) and 0.7 (n = 6) μg/kg per 24 hours were tested in the present study. We also investigated two doses of cGMP analogue [8-Bromo-cGMP (membrane-permeable), Sigma Chemical Co.], 0.1 (n = 5) and 0.3 (n = 5) mg/kg per 24 hours, as based upon the previous reports (Chi et al., 1999; Sarker and Fraser, 2002). Saline (0.9% NaCl, n = 6) was used as control vehicle.
Sampling and assessment (series 1)
Animals in series 1 were given an overdose of halothane (5%) to obtain a blood sample from the heart 24 hours after injury and then killed by decapitation to remove the brain. Normal values for all variables measured were obtained from five rats that were given an overdose of halothane without any injury or treatment (normal group in series 1, Table 1). The blood sample was used to measure plasma concentrations of sodium and potassium by means of flame photometry (943; Instrument Laboratory, Milan, Italy).
The water content of brain tissue was measured by the wet and dry weight method. Cerebral tissue was divided with a blade into the right and left hemispheres along the anatomic midline. The wet weight of each hemisphere was measured using an electronic analytical balance. After drying the sample in an oven at 95°C for 5 days, the dry weight was obtained. The water content of each hemisphere was calculated using the following formula: water content (%) = (wet weight − dry weight)/wet weight × 100.
For measurement of brain sodium and potassium concentrations, the dried samples were ashed by heating for 24 hours at 400°C. The ash was then extracted with distilled water and the concentrations of sodium and potassium were determined using a flame photometer (943; Instrument Laboratory) with cesium as an internal standard.
Evaluation of BBB integrity (series 2)
The integrity of the BBB was evaluated by assessing the extravasation of Evans blue dye as described previously (Beaumont et al., 2000). Briefly, the animal of series 2 was reanesthetized 23 hours and 15 minutes after injury and underwent a cannulation in the femoral vein with polyethylene tubing (P.E. 50; Becton Dickinson and Company, Sparks, MD, U.S.A.). Subsequently, 2% solution of Evans blue dye (E515; Fisher Scientific, Fairlawn, NJ, U.S.A.) in 0.9% NaCl was administered intravenously over 1 minute in a dose of 7mL/kg and then allowed to circulate for 30 minutes before the animal was killed.
Twenty-four hours after injury, a thoracotomy was performed after an overdose of halothane. Brains were perfused with approximately 500 mL of saline via a catheter inserted into the left ventricle of the heart and rapidly removed. The cerebrum of the brain was cut with a blade into the right and left hemispheres along the anatomic midline. Each hemispheric section was placed separately in a precisely measured volume (2 mL) of formamide and allowed to soak for 48 hours at room temperature. The supernatant solution was transferred to a microcuvette, and the absorbance of each solution was measured against a pure formamide standard at 625 nm using a Shimadzu UV 1600 Spectrophotometer (Shimadzu Instruments, Columbia, MD, U.S.A.). The tissue was then dried in the oven at 95°C for 5 days. Data was expressed as the relative absorbance (unit/g dry weight).
Normal values of the relative absorbance were obtained from five rats that were intravenously injected with Evans blue solution and killed 30 minutes later without any injury or treatment (normal group in series 2, Table 1).
Statistical analysis
All data are expressed as means ± SD. Statistical analyses were performed by one-way ANOVA followed by Fisher's post hoc test. Values of P < 0.05 are considered significant. Analyses were performed using the StatView Version 5.0 statistical package (Abacus Concepts, Berkeley, CA, U.S.A.).
RESULTS
Brain water content
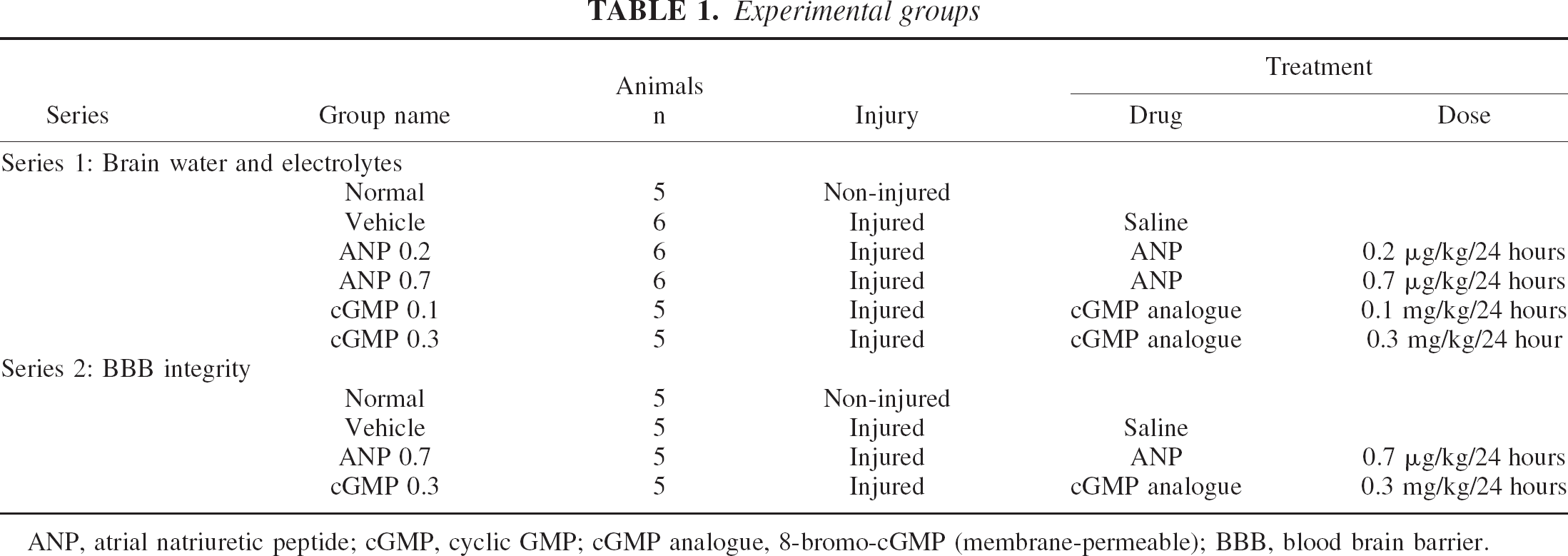
ANP administration exacerbated brain edema formation in the injured (right) hemisphere in a dose-dependent manner (Fig. 1). At 24 hours after injury, the brain water content in the injured hemisphere was significantly higher in ANP 0.2 (80.76 ± 0.26%, n = 6, P < 0.01) and 0.7 (80.86 ± 0.40%, n = 6, P < 0.002) groups than in the vehicle group (80.35 ± 0.32%, n = 6).
ANP effects on brain edema formation following the experimental cortical contusion. As compared with vehicle, ANP administration significantly exacerbated brain edema formation in the injured (right) hemisphere in a dose-dependent manner. ∗ P < 0.01; †P < 0.002, as compared with the vehicle group.
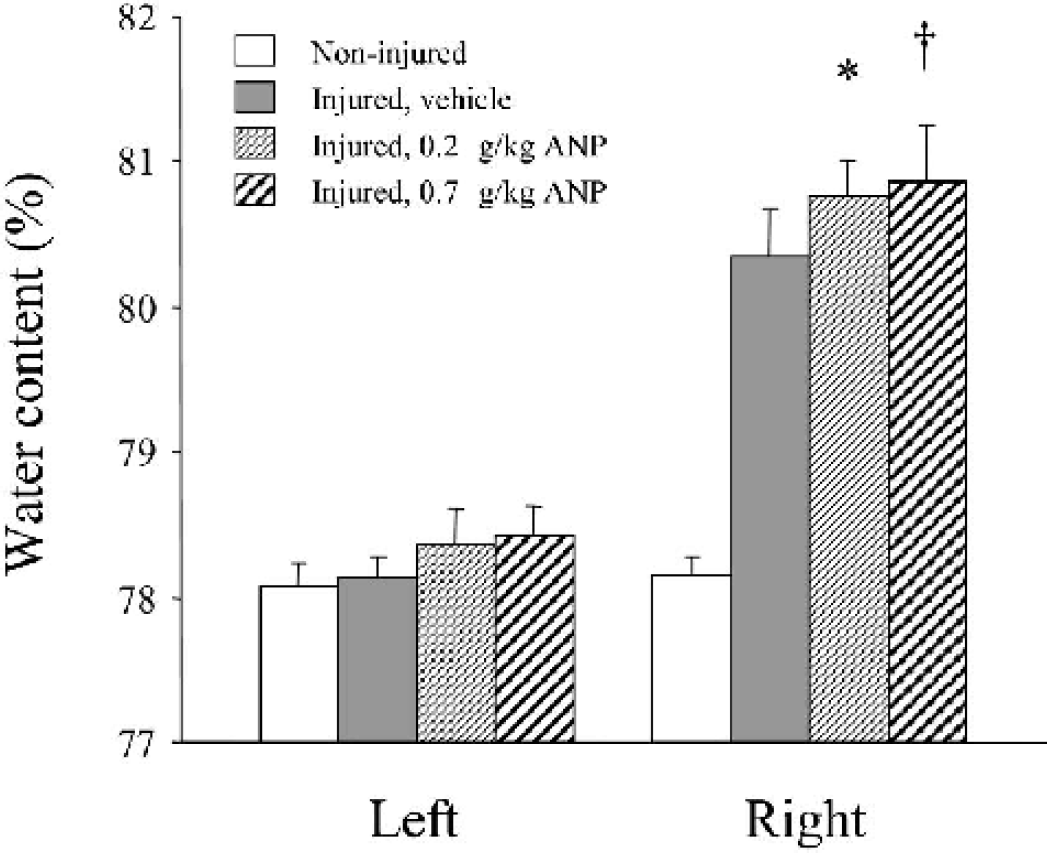
The cGMP analogue also exacerbated brain edema (Fig. 2). Both groups of cGMP 0.1 (80.80 ± 0.22%, n = 5, P < 0.002) and 0.3 (80.83 ± 0.20%, n = 5, P < 0.001) showed significantly higher water content in the injured hemisphere than did the vehicle group (80.35 ± 0.32%, n = 6).
The effect of cGMP analogue on brain edema formation following the experimental cortical contusion. As compared with vehicle, cGMP analogue significantly exacerbated brain edema formation in the injured (right) hemisphere in a dose-dependent manner. ∗ P < 0.002; †P < 0.001, as compared with the vehicle group.
Brain sodium level
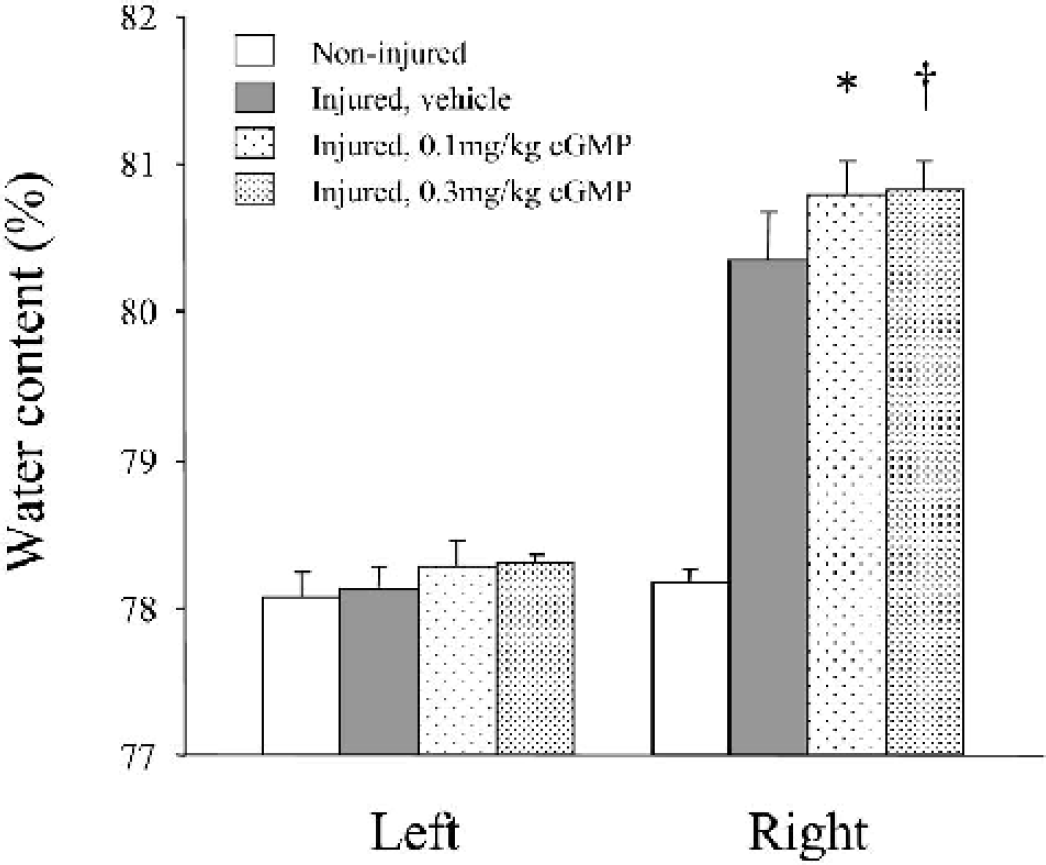
ANP administration reduced brain sodium concentrations in both hemispheres in a dose-dependent manner (Fig. 3). In the injured hemisphere, the brain sodium level was significantly lower in both groups of ANP 0.2 (305 ± 9 mEq/kg dry weight, n = 6, P < 0.05) and 0.7 (297 ± 26, n = 6, P < 0.01) than in the vehicle group (327 ± 19, n = 6). Similar effects were also observed in the noninjured (left) side, where the sodium level was significantly lower in ANP 0.2 (201 ± 18, n = 6, P < 0.02) and 0.7 (195 ± 14, n = 6, P < 0.005) groups, as compared with the vehicle group (226 ± 24, n = 6).
ANP effects on brain sodium concentrations following the experimental cortical contusion. As compared with vehicle, ANP administration significantly reduced brain sodium concentrations in both injured (right) and non-injured (left) hemispheres in a dose-dependent manner. ∗ P < 0.05; †P < 0.01; ∗∗ P < 0.02; ‡P < 0.005, as compared with the vehicle group.
The cGMP analogue also decreased the sodium level in both hemispheres (Fig. 4). The sodium level in the injured hemisphere was significantly lower in cGMP 0.1 (292 ± 5 mEq/kg dry weight, n = 5, P < 0.003) and 0.3 (281 ± 18, n = 5, P = 0.0002) groups than in the vehicle group (327 ± 19, n = 6). The sodium level in the noninjured side was also lower in both groups of cGMP 0.1 (190 ± 8, n = 5, P < 0.002) and 0.3 (185 ± 19, n = 5, P < 0.0005) than in the vehicle group (226 ± 24, n = 6).
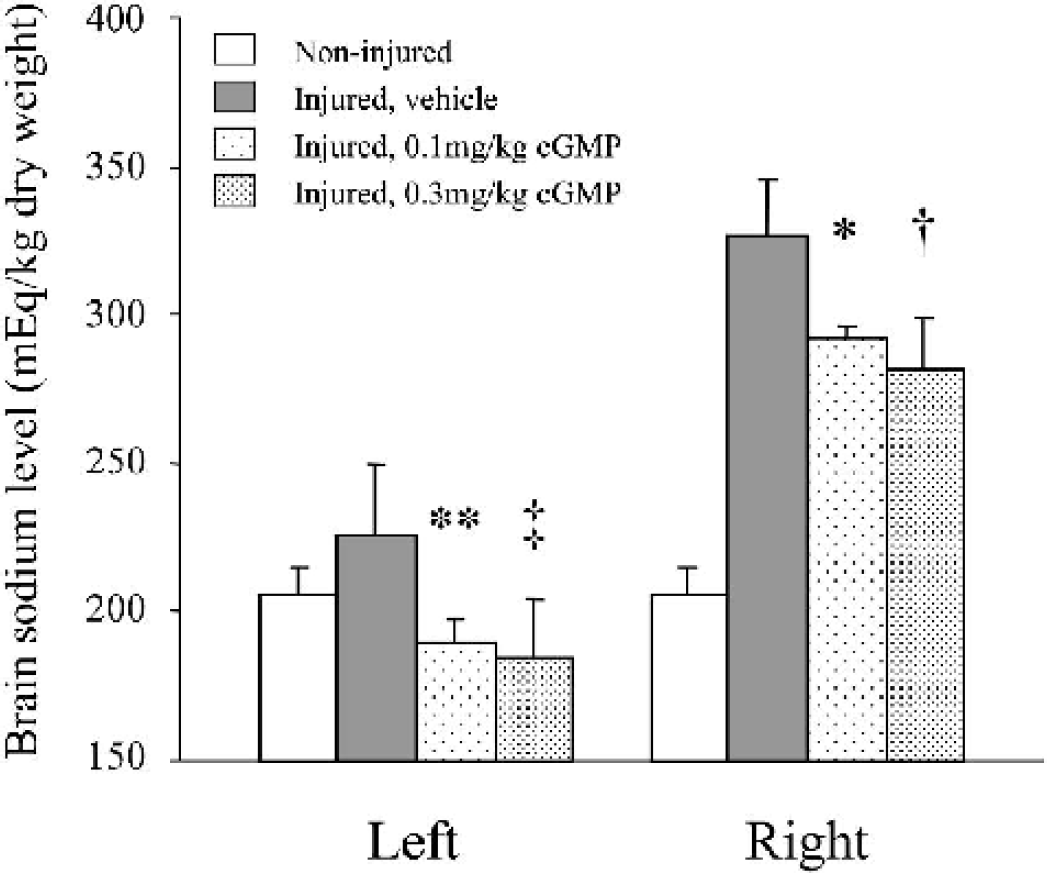
The effect of cGMP analogue on brain sodium concentrations following the experimental cortical contusion. As compared with vehicle, cGMP administration significantly reduced brain sodium concentrations in both injured (right) and non-injured (left) hemispheres in a dose-dependent manner. ∗ P < 0.003; †P = 0.0002; ∗∗ P < 0.002; ‡P < 0.0005, as compared with the vehicle group.
Brain potassium level
There was no significant difference in the brain potassium level between the vehicle and any drug group, whereas the potassium concentration of the vehicle group (311 ± 14 mEq/kg dry weight, n = 6) was significantly lower than that of the normal group (353 ± 16, n = 5, P = 0.003) in the injured hemisphere (Table 2).
The effect of ANP and cGMP analogue on the brain potassium level following the experimental cortical contusion
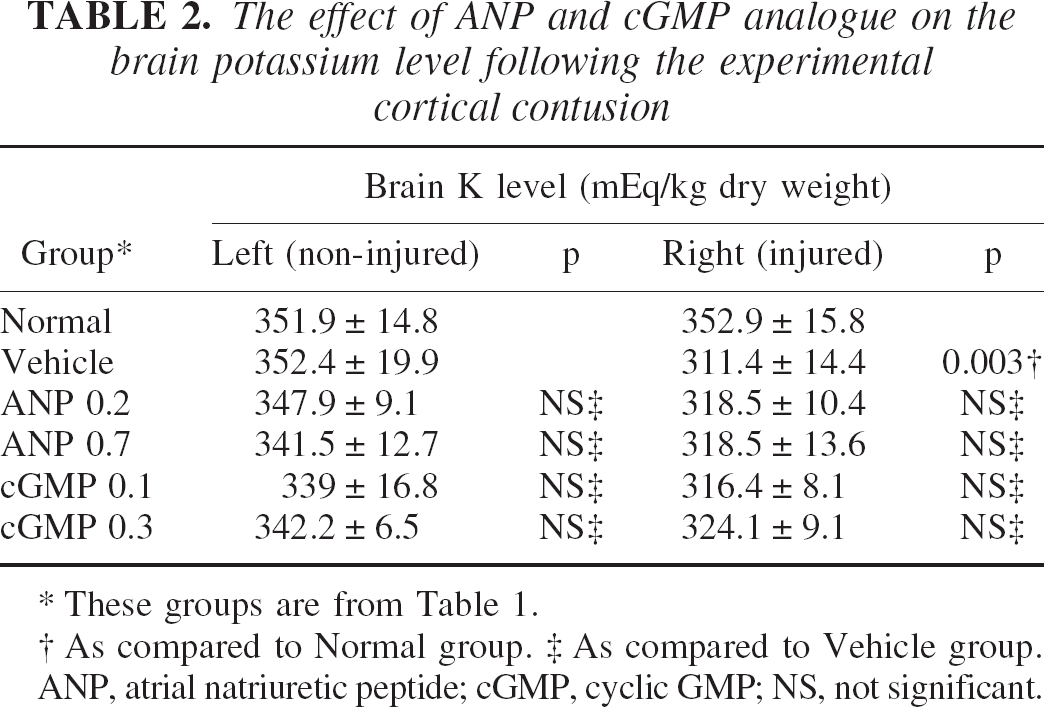
These groups are from Table 1.
As compared to Normal group.
As compared to Vehicle group.
ANP, atrial natriuretic peptide; cGMP, cyclic GMP; NS, not significant.
Plasma electrolytes level
Plasma electrolyte levels at 24 hours after injury are summarized in Table 3. There was no significant difference in the plasma level of either sodium or potassium between the vehicle and any drug group (Table 3).
The effect of ANP and cGMP on the plasma electrolytes level following the experimental cortical contusion
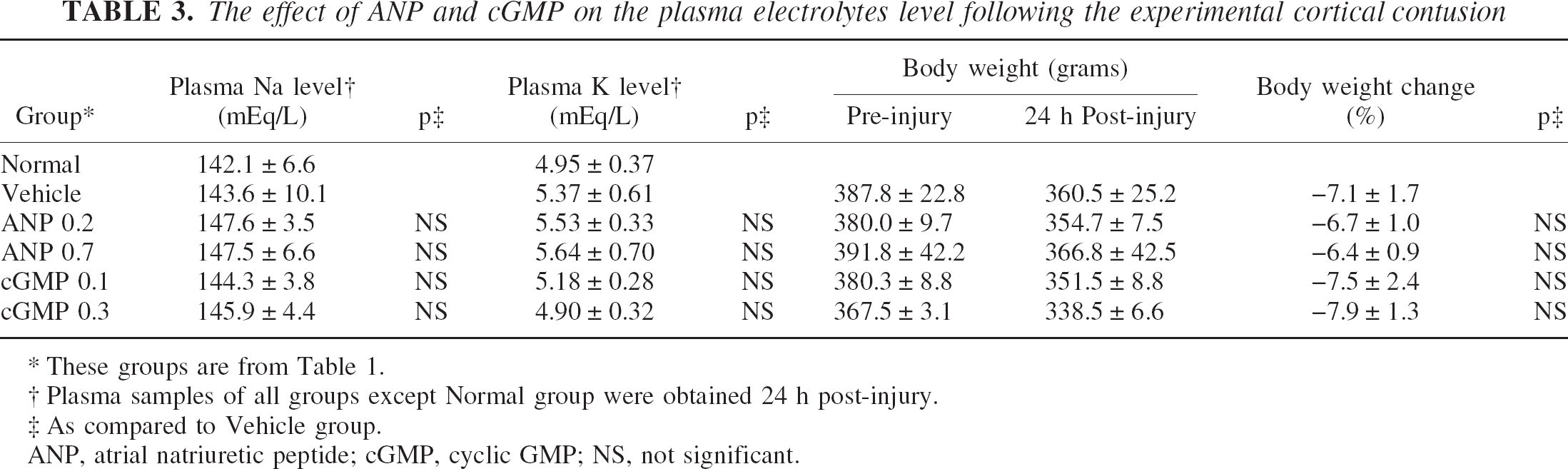
These groups are from Table 1.
Plasma samples of all groups except Normal group were obtained 24 h post-injury.
As compared to Vehicle group.
ANP, atrial natriuretic peptide; cGMP, cyclic GMP; NS, not significant.
Body weight change
All injured groups showed a reduction in the mean body weight at 24 hours after injury, with a range from −6.4 to −7.9% to each pre-injury weight. There was, however, no significant difference in the body weight change between the vehicle and any drug group (Table 3).
BBB integrity
Figure. 5 shows findings of the extravasation of Evans blue dye after the experimental cortical contusion. Extravasation of the dye was observed in the restricted area around the cortical contusion in the animal treated with vehicle (Fig. 5A). On the other hand, it was extensively observed in the injured (right) hemisphere in the animal treated with ANP 0.7 μg/kg/24 h (Fig. 5B) or cGMP analogue 0.3 mg/kg/24 h (Fig. 5C).
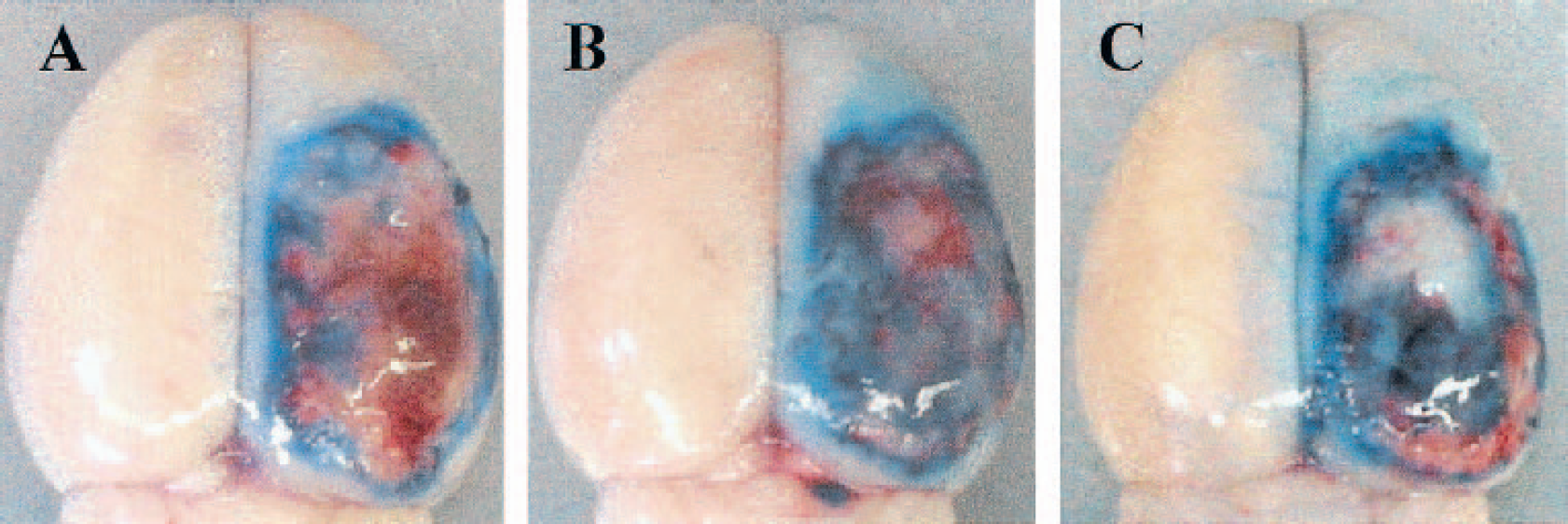
Demonstrable findings of the extravasation of Evans blue dye following the experimental cortical contusion. Extravasation of the dye was observed in the restricted area around the cortical contusion in the animal treated with vehicle
Semiquantitative analysis shows that the administration of ANP or cGMP analogue significantly exacerbated the extravasation in the injured hemisphere as compared with vehicle (Fig. 6). The relative absorbance in the injured hemisphere was higher in ANP 0.7 (3.21 ± 0.55 unit/g dry weight, n = 5, P < 0.01) or cGMP 0.3 group (3.53 ± 0.84, n = 5, P = 0.0004) than in the vehicle group (2.45 ± 0.56, n = 5) (Fig. 6).
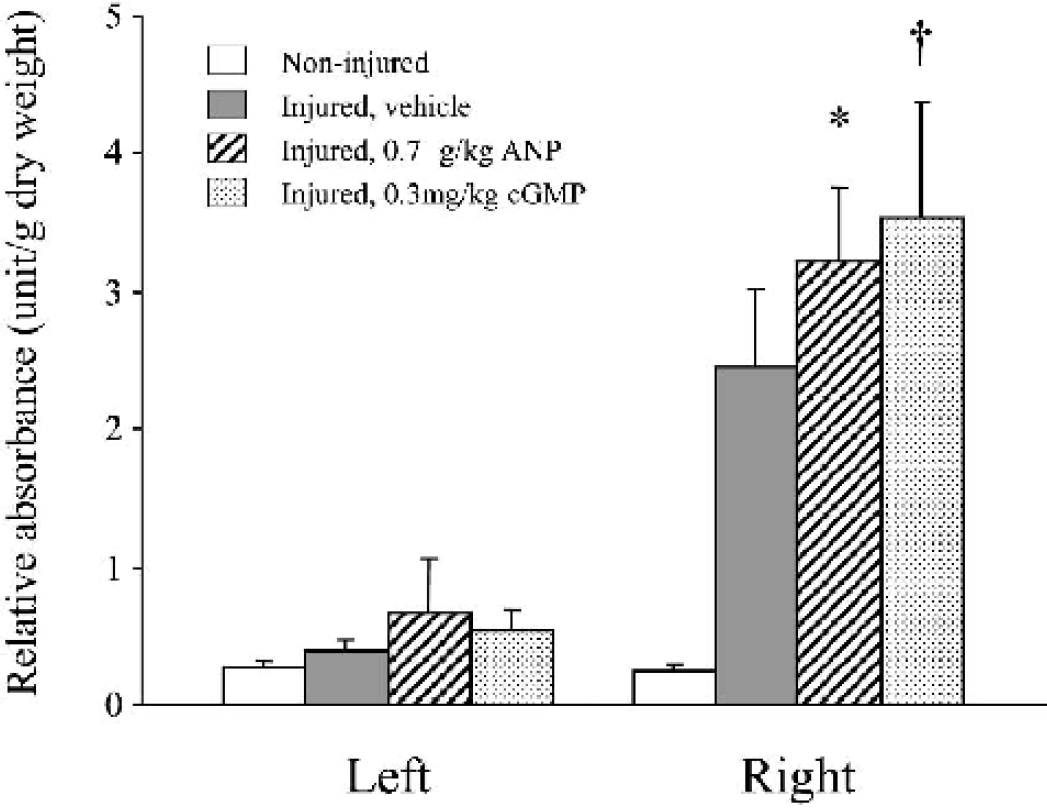
Semi-quantitative analysis of the extravasation of Evans blue dye following the experimental cortical contusion. Administration of ANP 0.7 μg/kg/24 h or cGMP analogue 0.3 mg/kg/24 h significantly exacerbated the dye extravasation in the injured (right) hemisphere than did vehicle. ∗ P < 0.01; †P = 0.0004, as compared with the vehicle group.
DISCUSSION
Interpretation of the results
In the present study, we suggest that ANP administration may have differential effects on brain water and sodium in TBI. In other words, ANP exacerbated brain edema formation after the experimental cortical contusion in rats, whereas it reduced brain sodium concentrations. These differential effects may appear contradictory as it is commonly believed that water and sodium tend to coexist and transfer together in the physiologic and pathologic state (Loo et al., 1996; Wright and Loo, 2000). We therefore attempted to elucidate the mechanism by which these effects could be mimicked by cGMP analogue. ANP administration exacerbated the BBB disruption as assessed by semiquantitative analysis of the extravasation of Evans blue dye. On the other hand, this peptide did not affect either plasma sodium levels or body weight as compared with vehicle. These findings suggest that this ANP effect on edema was attributed to an increase in the BBB permeability via cGMP pathway and not natriuresis or diuresis. It has recently been suggested that exogenous ANP may have little potential for natriuresis unless the extracellular fluid volume is high (Singh et al., 2002), thus possibly indicating that there might not be excessive water in the extracellular space in the present model. This is consistent with our previous experience that there is a major cellular component to the edema.
ANP effect on brain sodium
Recent evidence suggests that the luminal membrane of brain capillaries contains ANP receptors (Ermisch et al., 1991), which are coupled with guanylate cyclase (Steardo and Nathanson, 1987). The binding of ANP with these receptors activates a cyclic GMP (cGMP) (Light et al., 1989; Potter and Hunter, 2001). The activated cGMP then stimulates cGMP-dependent protein kinases (PKG), which have a potential for inhibiting an amiloride-sensitive sodium absorption (Ibaragi et al., 1989; Sarker and Fraser, 2002). Thus, ANP modulates unidirectional transport of sodium from blood to brain via the cGMP-PKG pathway by prohibiting amiloride-sensitive Na+/H+ exchange (Ennis et al., 1996; Ibaragi et al., 1989). We therefore speculate that this peptide may reduce brain sodium concentrations in brain injury by increases in another cation thereby contributing to the preservation of net cations in the brain. The nature of the Na depletion must be resolved in future studies.
ANP effect on brain edema formation
Recent evidence suggests that ANP reduces extravascular lung water in the dog model of oleic acid-induced pulmonary edema (Mitaka et al., 2002). This peptide also attenuated brain edema in the rat models of cerebral ischemia (Nakao et al., 1990; Naruse et al., 1991), intracerebral hemorrhage (Rosenberg and Estrada, 1995; Rosenberg et al., 1992), or induced hyponatremia (Vajda et al., 2001). Although it seems that these effects on tissue water can be explained by the modulation of sodium transport in peripheral or cerebral microvessels (Ennis et al., 1996; Ibaragi et al., 1989), one question has been raised as to why ANP administration exacerbated brain edema in the present model of cortical contusion differently than in other models.
As described previously, this peptide increases the permeability of BBB via cGMP-PKG pathway (Grammas et al., 1991; Sarker and Fraser, 2002; Vigne and Frelin, 1992). It is therefore reasonable that there are some differences in the ANP effect on tissue water in brains and other organs, such as lungs. However, the question still remains with regards to other brain injury models. One possible explanation is differences in the BBB integrity. In the present study, we used the CCI to induce an experimental cortical contusion in rats, thus leading to an increase in the BBB permeability as reported previously (Beaumont et al., 2000) or shown in the present study. ANP administration further exacerbated the BBB disruption. Consequently, this possible deleterious effect of ANP may require the preexistence of BBB disruption in the model.
The extent of BBB disruption in other models is unclear. In the first report concerning the ANP effect in cerebral ischemia (Nakao et al., 1990), an insult was induced by three vessel occlusion for 15 minutes, followed by a reperfusion of 15 or 30 minutes. Considering that BBB opens after several hours of ischemia (Dijkhuizen et al., 1999; Loubinoux et al., 1997), this model may have only a small disruption in the BBB. In a second study (Naruse et al., 1991), ANP was continuously administered, and the edema was evaluated 24 hours after the insult. Although this time course is similar to ours, there might be little disruption because an ischemic insult was induced by the permanent occlusion of middle cerebral artery. It is generally considered that permanent occlusion models mainly result in cellular edema, whereas transient occlusion models with reperfusion often cause vasogenic edema (Neumann-Haefelin et al., 2000; Yang and Betz, 1994). Brain edema found in the hyponatremic rat is cellular edema, not vasogenic, as assessed by the reduction of apparent diffusion coefficient (ADC) using magnetic resonance technique (Vajda et al., 2001). In the rat model of intracerebral hemorrhage, the timing and severity of BBB disruption may be influenced by the presence of lysed red blood cells (Bhasin et al., 2002). However, in the intracerebral hemorrhage model that was used to assess the ANP effect, bacterial collagenase was injected instead of blood (Rosenberg and Estrada, 1995; Rosenberg et al., 1992). Although there was one report suggesting BBB disruption in this model (Rosenberg et al., 1993), the mechanism of secondary brain edema still seems to be controversial.
Clinical implication
From the clinical point of view, it is important to know how ANP acts in TBI because this peptide has widely been used as preventive measures against heart failure (Mizuno et al., 2001), and severe TBI patients may often be associated with subsequent cardiac failure possibly because of a catecholamine discharge, apnea, or shock (Atkinson, 2000; Prichard et al., 1991). Thus, ANP may have been used in a percentage of TBI patients and will continue to be used unless other information about this possible deleterious effect is reported. We therefore suggest that ANP administration should not be given in patients with TBI until its effect on brain edema is also assessed in other models of TBI.
We have shown that ANP administration exacerbated brain edema in the rat model of cortical contusion. These effects could be mimicked by cGMP analogue, and ANP exacerbated the BBB disruption. These findings suggest that ANP effects on edema may be attributed to an increase in the BBB permeability via cGMP pathway. Further studies are, however, required to more thoroughly investigate the mechanism and determine if ANP administration is appropriate in patients with TBI.
Footnotes
Acknowledgment
The authors thank Virginia Godsey and Renee Glisson for their editorial assistance.