
Abstract
Transient forebrain ischemia induces a delayed neuronal death in the CA1 area of the hippocampus. However, the mechanism leading to this phenomenon has yet to be established. The authors used an mRNA differential-display method to isolate genes for which mRNA levels change only in the hippocampus during ischemia/reperfusion. They succeeded in identifying the product of one down-regulated gene as phosphatidylinositol 4-kinase (PI 4-K). Compared with control levels, PI 4-K mRNA expression in the hippocampus, but not the cerebral cortex, was significantly decreased by 30% and about 80% 1 and 7 days after ischemia/reperfusion, respectively. Interestingly, PI 4-K and PI bisphosphate levels were selectively decreased in the CA1 region, but not other regions, whereas TUNEL-positive cells could be detected 3 days after ischemia. Consistent with these results, PI 4-K expression was suppressed by hypoxia in SK-N-MC neuroblastoma cells before loss of cell viability. Overexpression of wild-type PI 4-K, but not the kinase-negative mutant of PI 4-K (K1789A), recovered the loss of viability induced by hypoxia. These findings strongly suggest that a prior decrease in PI 4-K and PI bisphosphate levels caused by brain ischemia/hypoxia is partly involved in delayed neuronal cell death.
Neurons in the striatum, hippocampus, and lateral thalamus die within a short period of global cerebral ischemia and after reperfusion in rodents (Kirino, 1982; Pulsinelli et al., 1982). In particular, pyramidal neurons in the CA1 region of the hippocampus undergo delayed neuronal cell death 3 to 5 days after transient forebrain ischemia (Kirino, 1982; Petito et al., 1987; Pulsinelli et al., 1982). Because it is considered that this period provides a therapeutic window after ischemic injury, there have been attempts to elucidate the exact mechanism of ischemia-induced neuronal cell death. Recently, evidence emerged that neurons are very fragile and sensitive to ischemic stress compared to glial cells and consequently undergo apoptotic cell death (Ferrer et al., 1994; Nitatori et al., 1995). The molecular mechanisms that trigger delayed neuronal cell death are not fully understood; however, hypotheses include the excitotoxicity of glutamate, a disturbed calcium homeostasis, an altered lipid metabolism, free radicals, and mitochondrial involvement (Abe et al., 1995; Juurlink and Sweeney, 1997). Furthermore, there have been several attempts to characterize the specific proteins up-regulated by brain ischemia such as heat-shock protein, transcriptional factors, neurotrophic factors, Bcl-2 family proteins, and caspases (Asahi et al., 1997; Chen et al., 1996, 1997; Choi, 1996; MacManus and Linnik, 1997; Jorgensen et al., 1989; Lindvall et al., 1992; Nowark, 1985). These observations led us to the conclusion that the proteins induced are at least partly involved in the promotion/attenuation of neuronal cell death.
The technique of mRNA DD has provided detailed information concerning changes in particular genes in response to brain ischemia in vivo. The method has been useful and in fact, several genes were isolated and identified successfully using ischemic brain. For example, in gerbil, a serine protease inhibitor gene (SPI-3) and zinc transporter gene (ZnT-1) were isolated from ischemic hippocampus with this method (Tsuda et al., 1996, 1997). In addition, adrenomedullin and the rat B2 sequence were discovered from rat cortex and hippocampus in a middle cerebral artery occlusion model and four-vessel occlusion model, respectively (Liu et al., 1999; Wang et al., 1995). These particular genes were up-regulated by each ischemic model and suggested to be involved in neuronal cell death.
In the present study, we attempted to further clarify the genes altered in the hippocampus, but not cerebral cortex, by brain ischemia in rats. From a screening by the DD method, we succeeded in isolating a specific down-regulated gene, never analyzed before. Our results indicated that PI 4-K is selectively down-regulated only in the CA1 subfield of the ischemic hippocampus. Because PI 4-K is a key enzyme in the production of PIP2 and PIP3, both of which play a critical role in cell survival signaling, the decrease in this enzyme may help to promote the delayed neuronal apoptosis in ischemic hippocampus.
MATERIALS AND METHODS
Materials
Anti-NeuN monoclonal antibody, anti–PI 4-K polyclonal antibody, and anti-PI4,5P2 monoclonal antibody (immunoglobulin M) were purchased from Chemicon, Santa Cruz, and Echelon, respectively. Anti-Akt and phospho-Akt polyclonal antibodies were obtained from New England Bio Labs. Anti–goat immunoglobulin G conjugated with Alexa 594, anti–mouse immunoglobulin G conjugated with Alexa 488, and streptavidin conjugated with Alexa 594 were purchased from Molecular Probes. MEBSTAIN Apoptosis Kit II was from MBL. Ampli Taq PCR polymerase and PLATINUM Pfx DNA polymerase were purchased from Perkin Elmer and Gibco BRL, respectively. All other reagents were obtained from Sigma.
Transient forebrain ischemia model
Adult male Wistar rats (250 to 300 g; Charles River Laboratories) were used for all of the experiments (Tanaka et al., 2000). All animal procedures conformed to the Medical Research Council of Japan's Guide for the Care and Use of Experimental Animals. Transient forebrain ischemia was produced by the 4VO method. Briefly, the bilateral vertebral arteries were electrocauterized and atraumatic clasps were placed around the common carotid arteries without interrupting the arterial blood flow. Two days later, forebrain ischemia was induced by tightening the clasps for 15 minutes. In the case of sham-treated animals, the carotids were exposed but not occluded. Body temperature was maintained at 37°C during and after the 4VO treatment using heat lamps. Animals were killed by decapitation at specific times after the ischemia or sham operation.
Total RNA isolation
The entire hippocampal and cerebral cortex regions from five rats were rapidly removed, frozen in liquid nitrogen, and stored at −80°C. Total RNA was extracted from brains of 4VO- and sham-treated animals by the acid guanidium/phenol/chloroform method.
Differential display
Initially, to remove contaminating DNA, the total RNA was incubated at 25°C for 40 minutes with RNase-free DNase (134 U/tube). The reaction was terminated by addition of edetic acid and sodium acetate, and RNA was phenol/chloroform-extracted and precipitated by 2-propanol.
For first-strand cDNA synthesis, total RNA (0.5 μg) was incubated with 1-μmol/L fluorescein isothiocyanate-conjugated GT15A, GT15G, and GT15C at 70°C for 10 minutes and then chilled on ice. Subsequently, the mixtures were incubated with 20-μmol/L dNTPs, 5-mmol/L DTT, 10 U RNase inhibitor, and 100 U Superscript II reverse transcriptase in 20 μL at 42°C for 1 hour and further incubated at 70°C for 10 minutes to terminate the reaction. Reverse transcriptase products were then used as PCR templates in 20 μL of reaction mixture containing random 10-mer primer (1-μmol/L), dNTP (200-μmol/L of each), and 2 U AmpliTaq polymerase or Pfx DNA polymerase. The PCR was performed for 30 cycles with a setting of 94 °C for 30 seconds, 40°C for 2 minutes, and 72°C for 1 minute (7 minutes for the last cycle). In one set of experiments, PCR products obtained from ischemic brains were screened using 30 different primers. After separation in 5% polyacrylamide gel electrophoresis, PCR products were stained with SYBR Green I (1:10,000) for 30 minutes and detected by FLA2000 (Fuji, Japan). cDNA bands that specifically changed in ischemic brains were cut out and eluted from the gels and reamplified by PCR using the same primers.
Cloning and sequencing of cDNA fragments
Reamplified cDNA fragments were cloned into pCR2.1-TOPO or pCR-Blunt II-TOPO (Invitrogen). Plasmid DNA sequencing of cloned fragments was carried out using the Perkin Elmer Gene Amp PCR system 2400 either with the T7 forward-sequencing primer or with RV13 primer. The cDNA sequences were analyzed and compared for homology with those available in EMBL and GenBank DNA databases.
Northern blot analysis
To confirm the reduction of PI 4-K transcript in response to ischemia/reperfusion, Northern blot analysis was performed using 5′-PI 4-K cDNA probe (1 to 1,108) amplified by PCR as described previously (Tanaka et al., 2000). Changes in PI 4-K mRNA were evaluated by comparison with β-actin mRNA.
Immunohistochemical analysis
At selected intervals after occlusion and reperfusion, rats were anesthetized with sodium pentobarbital (50 mg/kg, administered intraperitoneally), and the brain was removed and froze with liquid nitrogen. The frozen brains were cut with a cryostat into 10-μm coronal sections. The sections were washed with phosphate-buffered saline (PBS) for 10 minutes and then fixed with 4% formaldehyde for 20 minutes at room temperature. After a wash with PBS three times, the sections were incubated with 10% rabbit or goat normal serum for 1 hour. Then, the slices were incubated with first antibodies [anti-PI 4-K polyclonal antibody (1:200), anti-NeuN monoclonal antibody (1:200), and anti-PI4,5P2 monoclonal antibody (1:100)] at 4°C overnight. The sections exposed to the first antibodies were incubated with anti–mouse immunoglobulin G antibody conjugated with Alexa 488 (1:100), anti–goat immunoglobulin G antibody conjugated with Alexa 594 (1:100), and/or anti–mouse or biotinylated anti–mouse immunoglobulin M polyclonal antibody (1:400) for 1 hour at room temperature. In the case of the treatment with biotinylated anti–mouse immunoglobulin M polyclonal antibody, the sections were further incubated with streptavidin conjugated with Alexa 594 (1:200) at 37°C for 30 minutes. After a wash with PBS, the immunoreactive cells were visualized with Texas red and fluorescein. All images were taken on a Zeiss LSM 510 laser-scanning confocal microscope (Ko et al., 2002; Tanaka et al., 2000).
RT-PCR analysis
An aliquot (2 μL) of RT product was mixed with 2 U AmpliTaq polymerase and a 0.2-μmol/L concentration of each of the following sense and antisense primers in a final volume of 20 μL (Uehara et al., 1999a): rat PI 4-K, 5′-ATC CAG ATC GCT TCC AGG TAA-3′ (positions 2,934 to 2,954; upstream) and 5′-AAC GGC ACT TCC ACT CCA TTC-3′ (positions 3,931 to 3,951; downstream); rat PI 3-K (p85), 5′-TGT TGC CCC GCC TCT CCT TAT-3′ (positions 378 to 398; upstream) and 5′-ATT TCC TCC TTT CCT TAG TGT-3′ (positions 1,111 to 1,131; downstream); and rat GAPDH, 5′-AAA CCC ATC ACC ATC TTC CAG-3′ (positions 238 to 258; upstream) and 5′-AGG GGC CAT CCA CAG TCT CAG-3′ (positions 578 to 598; downstream).
Plasmids
Full-length rat 230-kd PI 4-K in pSRα was kindly provided by Prof. Kondo and Dr. Saino-Saito (Nakagawa et al., 1996). The PI 4-K expression vector with an HA tag fused to its N-terminal (pCR3.1-HA-PI 4-K) was engineered. A kinase negative PI 4-K (K1789A) mutant was generated by the overlapping PCR method and the cDNA was subcloned into pCR3.1 [pCR3.1-HA-PI 4-K (K1789A)].
Cell culture
Human neuroblastoma SK-N-MC cells were maintained in modified Eagle's medium supplemented with 10% (vol/vol) heat-inactivated fetal calf serum at 37°C in a humidified 5% carbon dioxide, 95% air atmosphere (Araya et al., 1998).
Exposure to a hypoxic environment
When the cells became subconfluent, they were cultured in a mixture of 5% carbon dioxide and the balance of nitrogen in a humidified incubator (ANX-1, HIRASAWA, Tokyo, Japan) at 37°C within a sealed, anaerobic, gloved cabinet containing a catalyst to scavenge free oxygen as described previously (Ko et al., 2002; Tanaka et al., 2000; Uehara et al., 1999a). The medium was then changed, and the cells were cultured under normal conditions for given periods (reoxygenation).
DNA fragmentation
For the DNA fragmentation assay, the cells were lysed in lysis buffer [10-mmol/L Tris-hydrochloride (pH 7.4), 5-mmol/L edetic acid, and 0.5% Triton X-100) and incubated for 20 minutes at 4°C. The samples were centrifuged at 27,000 g for 15 minutes at 4°C. The supernatants were incubated with 40-μg/mL proteinase K for 30 minutes at 37°C and extracted with equal volumes of phenol, phenol/chloroform (1:1 vol/vol), and chloroform. The DNA was precipitated from the supernatants with a 1/10 volume of 3 M sodium acetate (pH 5.2) and 2 volumes of ethanol and treated with 40 μg/ml of RNase A for 1h at 37°C. The recovered DNA was then analyzed by electrophoresis on a 1.5% agarose gel and visualized with 0.5-μg/mL ethidium bromide (Uehara et al., 1999b).
Assessment of cell viability
Viability was measured by the counting of viable cells stained for β-galactosidase activity derived from transfection of that gene (Ko et al., 2002; Tanaka et al., 2000).
RESULTS
Identification of clone a-15
In this study, we focused on the genes that are selectively up- or down-regulated in the hippocampus, not the cerebral cortex, because a typical delayed neuronal apoptosis was observed only in the former. The products from each DD-PCR were loaded in duplicate. Fig. 1 shows a typical result of DD-PCR amplified by using a random primer (gat cca gta c) and an anchored oligo(dT) primer. The PCR products were subjected to gel electrophoresis and then stained with SYBR Green in order to detect the specific bands. In this fingerprint, several specific bands that increased or decreased only in the hippocampus after ischemia/reperfusion were detected (Fig. 1A). In particular, the clone designated as a-15 was detected in both the cerebral cortex and the hippocampus of sham-operated rats in quiescent states. Interestingly, the transcript had almost disappeared 1 day after ischemia/reperfusion in the hippocampus, whereas the level was unchanged during this period in the cerebral cortex. Sequence analysis revealed that the transcript completely coincided with that of the 3′-terminal of the 230-kd PI 4-K (6,484 to 6,744) (Nakagawa et al., 1996). In this region, the random primer and anchored oligo(dT) primer used were certainly included (Fig. 1B). To ascertain whether the findings are reproducible, we used the RT-PCR method to detect PI 4-K using another set of primers. Fig. 1C shows that PI 4-K expression is selectively attenuated in response to ischemia/reperfusion in the hippocampus, but not the cerebral cortex, as seen in the results of DD-PCR. Subsequently, we investigated whether PI 3-K, one of the PI kinases, is also decreased by brain ischemia in hippocampus. However, a significant decrease in PI 3-K (p85 subunit) mRNA was not observed during 7 days of ischemia/reperfusion in either area. These results suggest that the decrease in PI 4-K in hippocampus caused by brain ischemia is a specific phenomenon (Fig. 1C). However, we have identified that the rat B2 sequence, which has been already reported, was transiently up-regulated, peaking at 1 day after brain ischemia in hippocampus but not cerebral cortex (data not shown) (Liu et al., 1999).
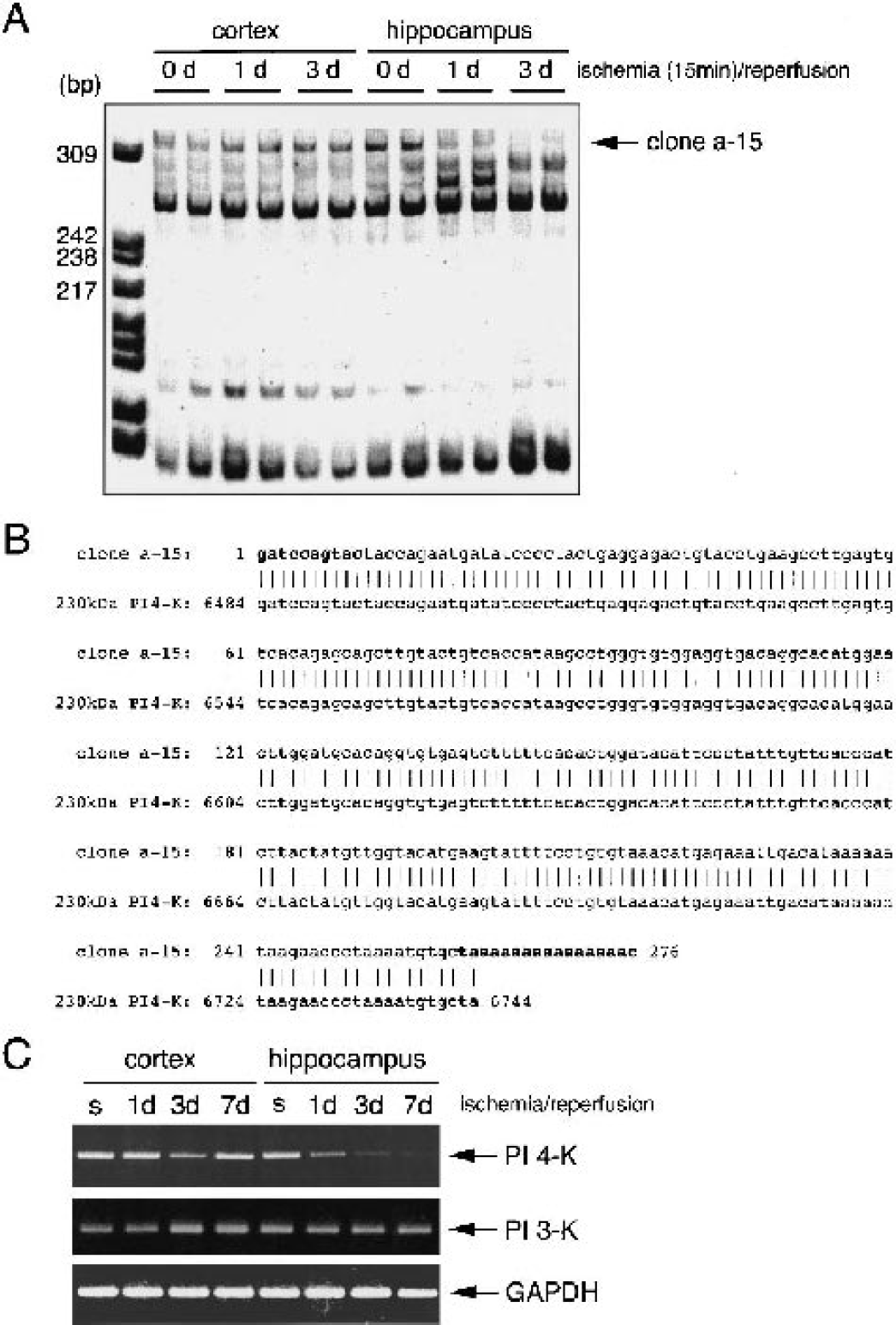
Isolation and identification of a particular gene, which is down-regulated by transient forebrain ischemia in hippocampus, but not cerebral cortex.
Specific down-regulation of PI 4-K in the CA1 region of hippocampus by brain ischemia
To confirm the gene-expression patterns observed in the DD analysis, the PI 4-K fragment (1 to 1,101) was used as a probe. Kinetic analysis revealed that PI 4-K mRNA levels in hippocampus decreased to approximately 30%, 60%, and 75% of the control values 1, 3, and 7 days after ischemia/reperfusion, respectively (Fig. 2A and Fig. 2B). To ascertain the regions of the decrease in hippocampus and whether the reduction of PI 4-K mRNA caused by brain ischemia induces the down-regulation of PI 4-K protein expression, we conducted immunohistochemical analyses on brain slices using anti-PI 4-K antibody and anti-NeuN antibody (which recognizes neuron-specific nuclear protein). In addition, we simultaneously investigated the DNA-fragmented cells detected by in situ end labeling using other independent samples. On indirect immunofluorescence labeling of PI 4-K, the antigen (green) was mainly observed in pyramidal neurons and to some extent in other types of cells (maybe glia) of the hippocampus (Fig. 3A and Fig. 3B, data not shown). The magnified images showed that the strong intensity (red signal) based on PI 4-K immunoreactivity is detected in cytoplasm containing cell membrane. NeuN, a neuron-specific protein, was detected in nuclei of pyramidal cells (green signal). Merged images indicated that the signal of PI 4-K reactivity is quite different from that of NeuN, although the yellow signal (both PI 4-K–positive and NeuN-positive localization) was partly seen. The immunoreactivity began to attenuate 12 hours after brain ischemia, had almost disappeared 1 day after ischemia, and was completely undetectable 3 days after ischemia in the CA1 subfield of the hippocampus (Fig. 3A). However, a significant number of TUNEL-positive cells were detected 3 days after brain ischemia only in the CA1 subfield, with this number increasing markedly thereafter. In the dentate gyrus of the hippocampus, there were no significant changes in the immunoreactivity for anti-PI 4-K and anti-NeuN antibodies. Furthermore, TUNEL-positive cells were undetectable in this area (Fig. 3B).
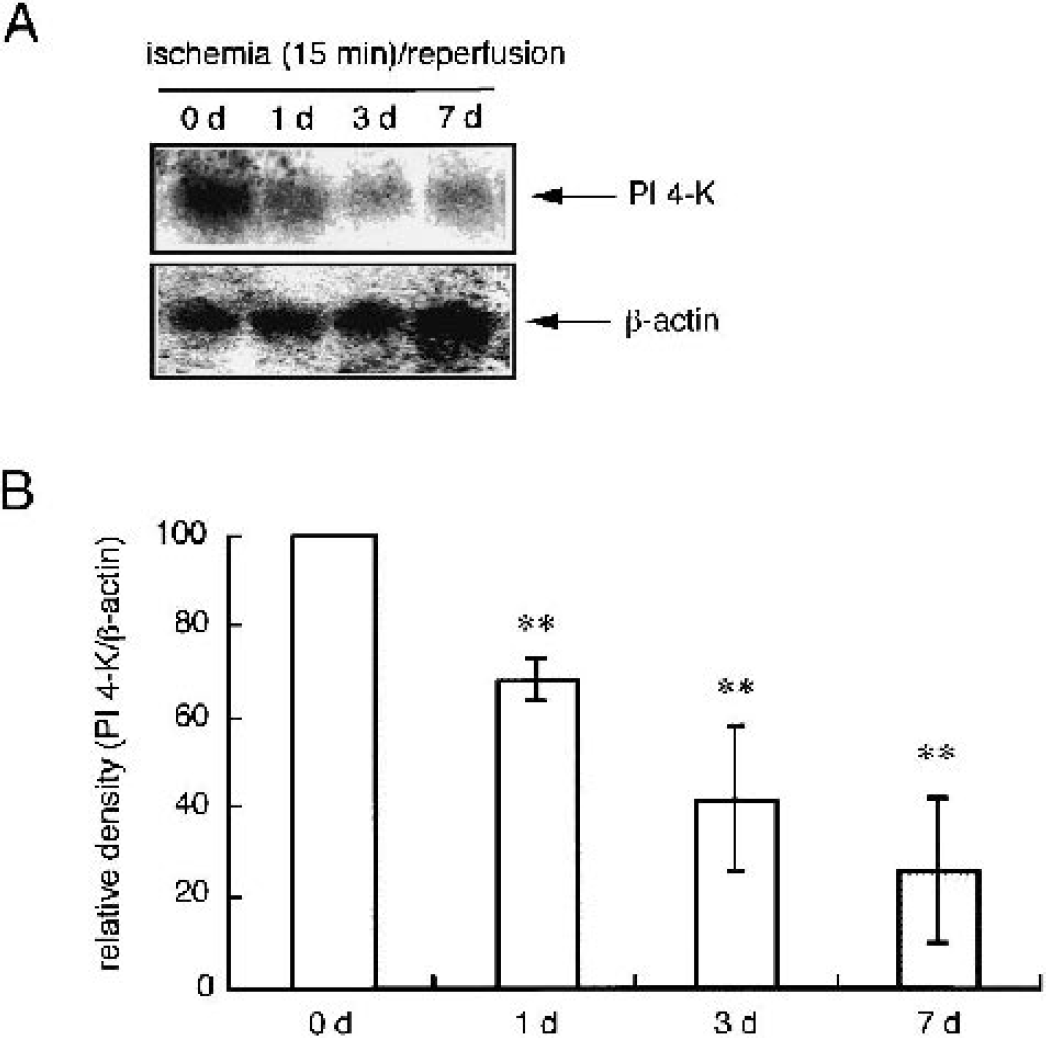
Expression levels of hippocampal phosphatidylinositol 4-kinase (PI 4-K) transcripts during the ischemic insult.
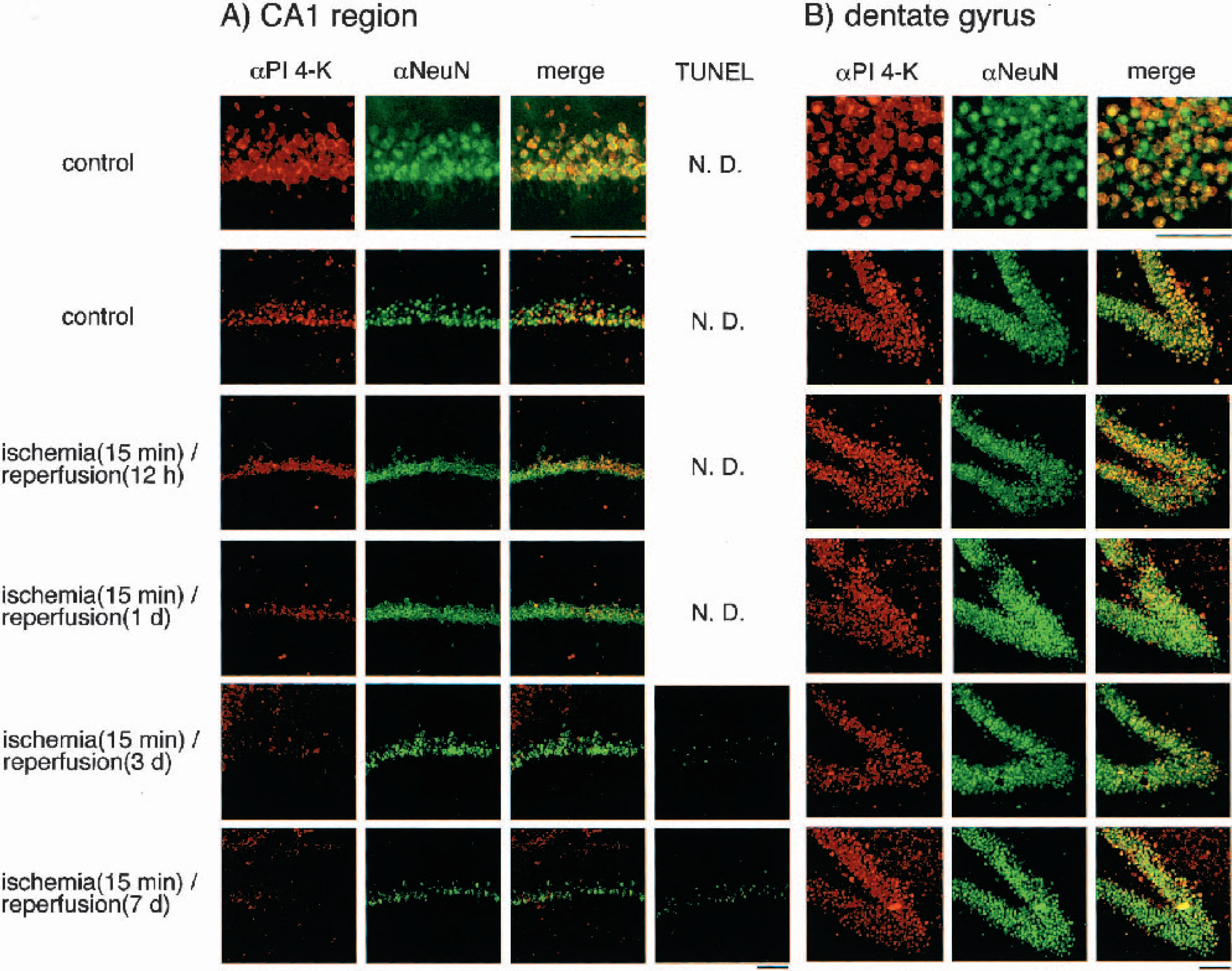
Changes in phosphatidylinositol 4-kinase (PI 4-K) levels during ischemia/reperfusion in the CA1 region and dentate gyrus of rat hippocampus. Confocal microscopic images indicating the localizations and levels of PI 4-K (red signal)/NeuN (green signal) immunoreactivity
Changes in PI(4,5)P2 levels caused by brain ischemia
We next investigated the changes in PI(4,5)P2 levels in hippocampus during brain ischemia. In the hippocampus, PI(4,5)P2-positive cells were mostly detected in the pyramidal cell layer. As seen in the results of immunohistochemical analysis with anti PI 4-K antibody, the immunoreactivity for anti-PIP2 antibody began to decrease 1 day after ischemia, and was hardly detectable 3 days after ischemia in the CA1 subfield of the hippocampus (Fig. 4A). However, in the dentate gyrus of the hippocampus, there were no significant changes in immunoreactivity for anti-PI 4-K and anti-NeuN antibodies (Fig. 4B).
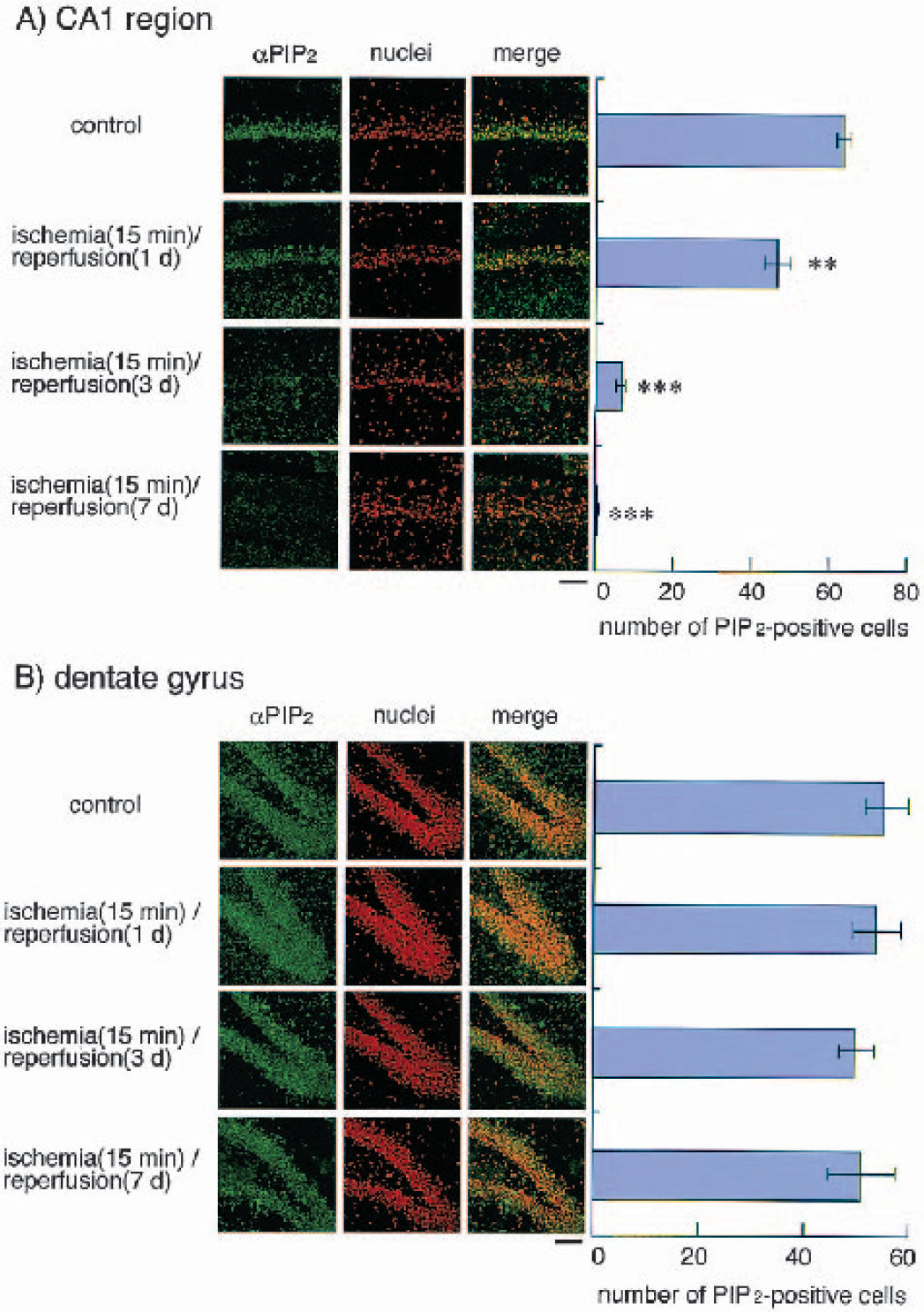
Detection of phosphatidylinositol bisphosphate (PIP2) levels during ischemia/reperfusion in the CA1 region and dentate gyrus of hippocampus. Left panels, confocal microscopic images indicating the levels of PIP2 (green signal) and nuclei staining (red signal) with propidium iodide
Down-regulation of PI 4-K in neuronal cells by hypoxia in vitro
We attempted to elucidate whether a decrease in PI 4-K is observed after treatment with hypoxia (one of the major types of ischemic stress) during neuronal apoptosis in vitro. Fig. 5 shows the changes in PI 4-K levels with hypoxia treatment in human neuronal SK-N-MC cells. Whole-cell lysates were prepared from the cells exposed to hypoxia for the indicated periods and were then subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis and Western blotting analysis. The levels were decreased after 6 hours in response to hypoxia and all PI 4-K disappeared after 18 hours.
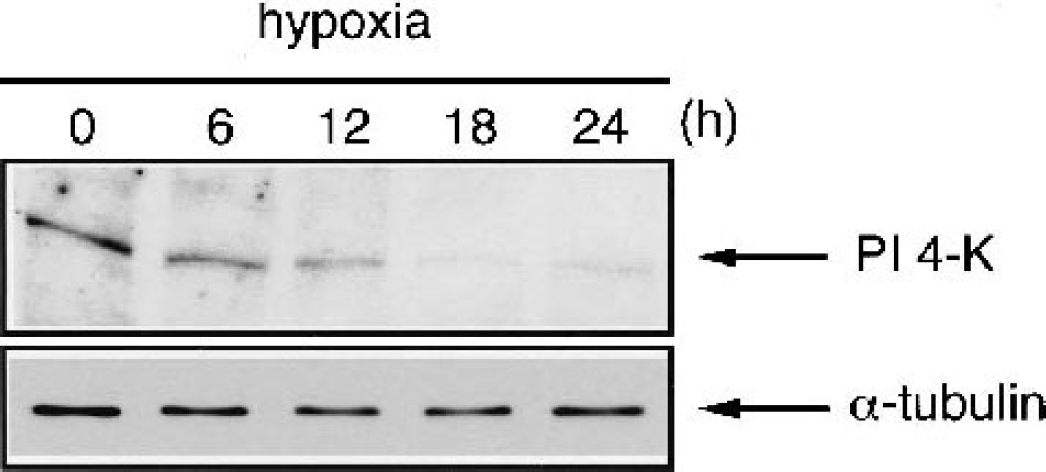
Decrease in phosphatidylinositol 4-kinase (PI 4-K) levels in response to hypoxia in human neuroblastoma SK-N-MC cells. Total cell lysates were prepared from cells exposed to hypoxia for various periods and then subjected to Western blotting analysis using anti-PI 4-K antibody. Results of the kinetic analysis of the levels of PI 4-K are shown.
Effects of overexpression of wild-type and kinase-inactive PI 4-K on the loss of cell viability by hypoxia
Recently, it has been reported that Lys-1792 in bovine brain 230-kd PI 4-K has a critical residue for kinase activity (Vereb et al., 2001). This Lys residue in bovine PI 4-K is equivalent to Lys-1789 in human PI 4-K (Fig. 6A). To confirm the role of PI 4-K in neuronal cell death in vitro, we investigated the effects of this molecule on hypoxia-induced cell death. To address the ability of wild-type or kinase-negative mutated PI 4-K (K1789A) to protect against hypoxia-induced cell death, we enumerated living β-galactosidase–positive cells. The transient expression of wild-type PI 4-K resulted in a significant recovery of cell viability by treatment with hypoxia for 21 hours compared with the mock transfectant (Fig. 6B). However, the K1789A mutant showed no alteration in the loss of cell viability. Immunohistochemical analyses revealed that overexpression of wild-type PI 4-K enhances the immunofluorescent intensity toward anti-PI(4,5)P2 antibody compared with the mock transfectant (Fig. 6C). In addition, immunofluorescent staining of PI(4,5)P2 revealed cell-surface staining and the pattern was reminiscent of cell membrane staining, suggesting that overexpression of PI 4-K enhances the PI(4,5)P2 levels in cell membranes. However, the immunofluorescent intensity for anti-PI(4,5)P2 antibody decreased to undetectable levels on treatment with hypoxia for 21 hours in the mock transfectant, overexpression of PI 4-K prevented cell death and significantly detectable levels of immunofluorescent intensity were maintained at that time.
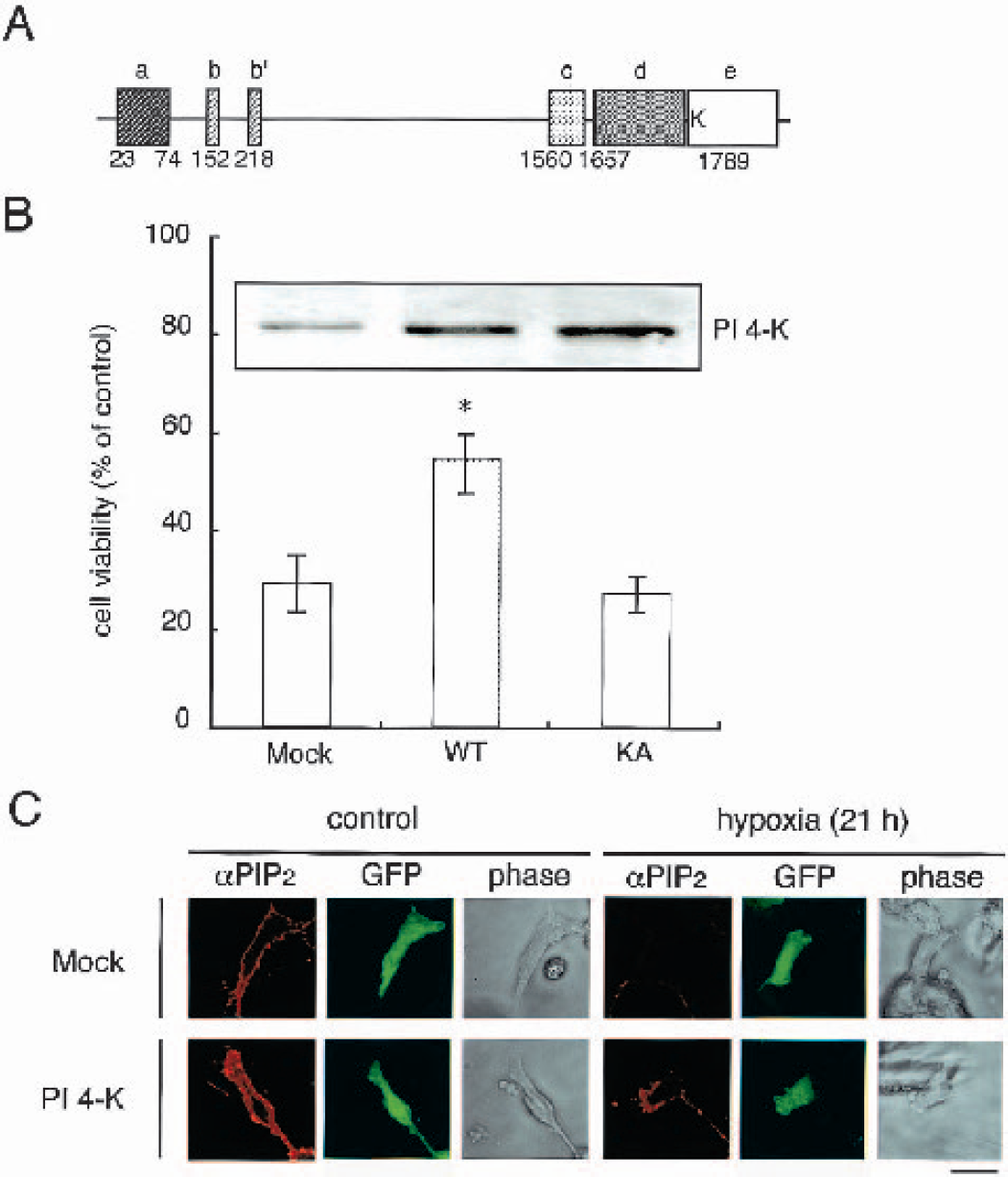
Overexpression of phosphatidylinositol 4-kinase (PI 4-K) protects against the loss of cell viability induced by hypoxia in SK-N-MC cells.
Effect of PI 4-K overexpression on Akt activation by hypoxic treatment
PI 4-K participates in the production of phosphoinositides such as PIP and PIP2, and is therefore a key enzyme for the production of PI(4,5)P2, which is a substrate for PI 3-K. Production of PI(3,4)P2 and PI(3,4,5)P3 through PI 3-K activates the Akt/PKB pathway, which results in the inhibition of apoptosis. Hence, we investigated the activation of Akt by hypoxia in mock- and PI 4-K–transfected cells. In mock-transfected cells, hypoxic treatment transiently stimulated Akt phosphorylation, which then peaked at 18 hours. However, the activation of Akt evoked by hypoxia in PI 4-K–transfected cells was sustained for up to 24 hours (Fig. 7). In addition, we examined the effect of PI 3-K inhibitor on attenuation of hypoxia-induced DNA fragmentation by PI 4-K in neuronal cells. Fig. 8 shows that hypoxia results in the formation of a DNA ladder after 30 hours in human neuroblastoma SK-N-MC cells, and overexpression of PI 4-K partially attenuated this formation. Treatment with wortmannin, a PI 3-K inhibitor, exacerbates the suppression of hypoxia-induced DNA fragmentation by PI 4-K in neuronal cells.
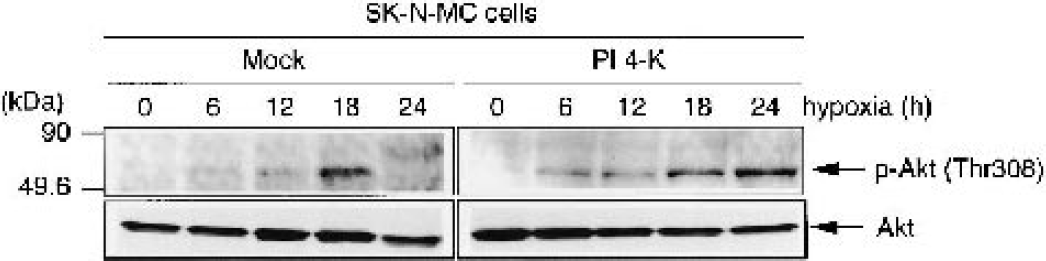
Overexpression of phosphatidylinositol 4-kinase (PI 4-K) enhances phosphorylation of Akt at threonine 308 induced by hypoxia. Cells were transfected with 2.0 μg of the vector (pCR3.1 or wild-type PI 4-K). Thirty-six hours after transfection, each transfectant was transferred to a low-oxygen chamber and incubated for the periods indicated. Then, total-cell lysates were prepared and subjected to Western blotting analysis using anti—phospho-specific (Thr308) Akt or Akt antibody.
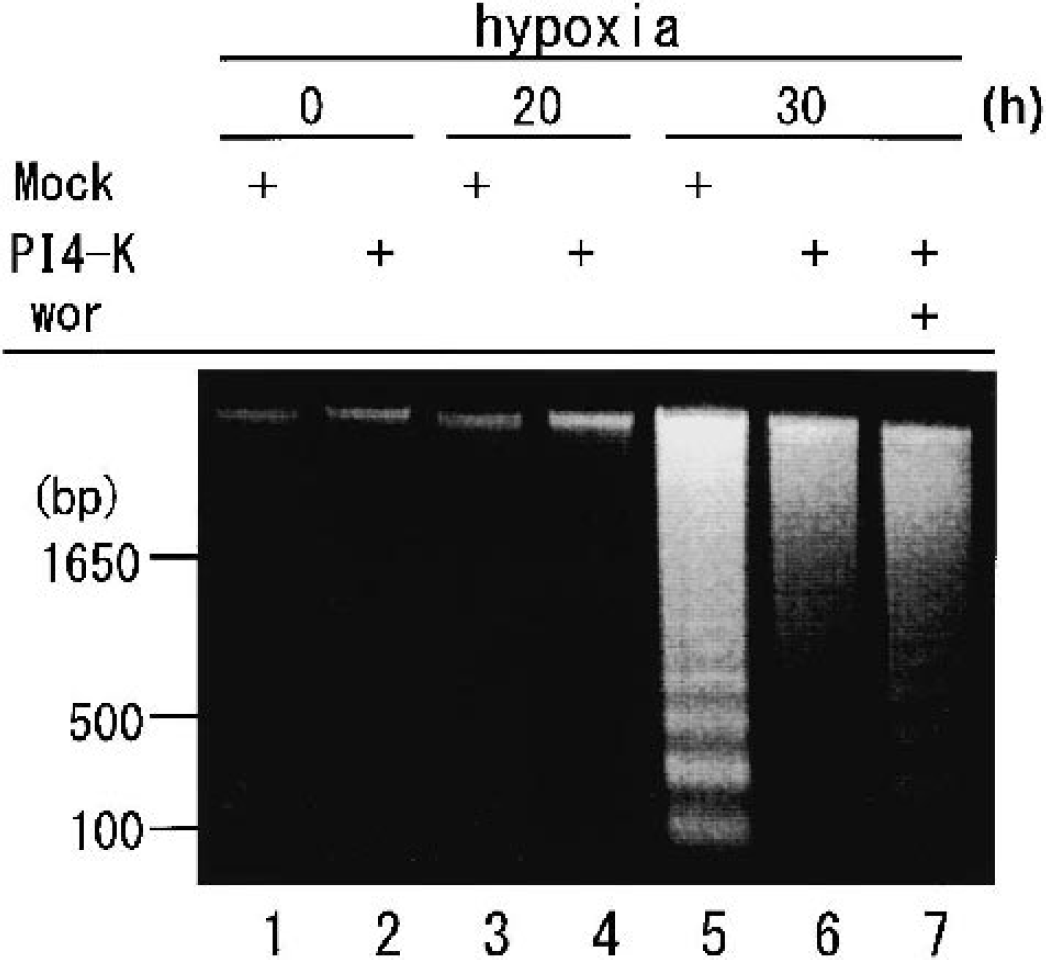
Treatment with wortmannin exacerbates the suppression of hypoxia-induced DNA fragmentation by phosphatidylinositol 4-kinase (PI 4-K). SK-N-MC cells were transfected with 2.0 μg of the vector (pCR3.1 or wild-type PI 4-K). Thirty-six hours after transfection, each transfectant was transferred to a low-oxygen chamber and incubated for the periods indicated with or without 100-nmol/L wortmannin. Then, total cell lysates were prepared and subjected to a DNA-fragmentation assay.
DISCUSSION
Isolation and identification of PI 4-K
The purpose of this study was to isolate and identify the genes implicated in the delayed neuronal cell death caused by transient forebrain ischemia in rats. We used the mRNA DD method to isolate those genes for which levels change with brain ischemia. We established the following criteria for this screening: the change in a candidate gene or gene product during brain ischemia was observed only in hippocampus, not cerebral cortex, only in the CA1 subfield, not in other areas such as the dentate gyrus, and at 1 day after ischemia/reperfusion, because the delayed neuronal apoptosis is detected 3 days after ischemia. On the basis of these criteria, we have succeeded in isolating two genes through DD analysis. One is identical to the B2 sequences recently reported as an up-regulated gene (Liu et al., 1999). Unfortunately, the B2 sequences protein has not been detected yet; however, it is speculated to be involved in gene expression and RNA processing (Krayev et al., 1982). The other is identical to 230-kd PI 4-K (Nakagawa et al., 1996). Interestingly, the expression of PI 4-K mRNA was significantly down-regulated in response to brain ischemia. To date, numerous genes and gene products that could be involved in delayed neuronal cell death have been found and characterized. The candidates reported are almost all up-regulated. Because there has been little attempt to identify selective down-regulated genes, we focus on the role of PI 4-K, the expression of which is decreased by brain ischemia.
Changes in PI 4-K protein PIP2 content in ischemic hippocampus
As shown in Fig. 2A and Fig. 2B, Northern blotting analysis revealed that the level of PI 4-K mRNA was already decreased at 1 day after ischemia and gradually declined thereafter. Thus, we investigated whether PI 4-K immunoreactivity is also decreased consistent with the change in mRNA, and which area of the hippocampus is influenced by ischemia. Initially, PI 4-K immunoreactivity was detected mostly in the pyramidal cells of the CA1, CA2, CA3, and dentate gyrus subfields in control rats (Fig. 3A and Fig. 3B, data not shown). Surprisingly, the intensity toward PI 4-K antigen began to decrease at 1 day after ischemia/reperfusion only in the CA1 region, whereas TUNEL-positive cells in this area, but not the dentate gyrus, were detected 3 days after ischemia/reperfusion. These results indicated that the phenomenon occurs selectively in the CA1 subfield and before delayed neuronal death. PI 4-K is an enzyme that phosphorylates PI or PIP and consequently produces PIP or PIP2. Therefore, we speculated that PIP2 levels in PI 4-K–positive cells are also decreased during ischemic insults. Consistent with this hypothesis, the immunohistochemical analysis with anti-PIP2 antibody strongly indicated that PIP2-positive cells were also decreased at 1 day after ischemia/reperfusion in the CA1 subfield, but not the dentate gyrus of the hippocampus (Fig. 4). From these observations, we concluded that transient forebrain ischemia results in the reduction of PI 4-K mRNA and consequent PIP2 levels before delayed neuronal cell death in the CA1 subfield of hippocampus in rats.
Decrease in PI 4-K during hypoxia-induced neuronal apoptosis
We subsequently determined whether a reduction of PI 4-K occurred during neuronal apoptosis induced by hypoxia, one of the ischemic stresses, in vitro. Human neuroblastoma SK-N-MC cells undergo apoptosis 24 hours after hypoxic treatment (Araya et al., 1998). Levels were significantly decreased after 6 hours in response to hypoxia, and no PI 4-K was detected after 18 hours. These findings suggested that the decrease in PI 4-K in the neuronal cell line occurs before detection of typical apoptotic features. To clarify the possible function of PI 4-K in hypoxia-induced apoptosis, we investigated the effect of the enzymic activity through overexpression into neuronal cells. Transfection of wild-type PI 4-K rendered the cells more resistant to the damaging effects of hypoxic treatment (Fig. 6B). At that time, the immunoreactivity of PIP2 was maintained during hypoxic treatment in PI 4-K-transfected cells, compared with that in mock-transfected cells (Fig. 6C). However, the kinase-negative PI 4-K mutant could not confer resistance to hypoxia, suggesting that the kinase activity of PI 4-K is critical for protection against hypoxic stress. Furthermore, the loss of PI 4-K due to brain ischemia and hypoxia in neuronal cells may lead to apoptotic cell death.
PI 4-K is implicated in the production of PIP or PIP2 and is a key enzyme for the subsequent production of phosphoinositides such as PIP3. In general, PI 3-K, which is stimulated by growth factors, is involved in the production of PIP3 that activates the Akt/PKB pathway by using PIP or PIP2 as a substrate. Akt/PKB is well-known to be involved in cell-survival signaling and, in fact, could attenuate some apoptotic events. Therefore, PI 4-K may have an important role in endogenous cell-survival or antiapoptotic signaling. Recently, it was reported that hypoxia triggers a cell-survival pathway through PI 3-K/Akt in PC12 cells (Alvarez-Tejado et al., 2001). In our system, hypoxia transiently stimulated a peak in Akt/PKB at 18 hours (Fig. 7). This activation was sensitive to wortmannin and LY294002 (PI 3-K inhibitors), suggesting that PI 3-K is mediated by hypoxia-induced Akt/PKB activation (data not shown). It seems that hypoxia simultaneously stimulates cell-death and cell-survival pathways. In these neuronal cells, the death-signal pathway induced by hypoxia may be more potent than the survival pathway, because treatment with PI 3-K inhibitors accelerated the appearance of apoptotic-sensitive features (Fig. 8). However, phosphorylation of Akt was observed in PI 4-K–transfected neurons in a sustained manner (Fig. 7). From these observations, we speculate that overexpression of PI 4-K helps to maintain the protein and PIP2 levels against the decrease induced by hypoxia and to sustain the activation of the cell-survival pathway involving PI 3-K/Akt induced by hypoxia, thereby ultimately contributing to the survival of neurons.
Possible roles of PI 4-K in the CA1 subfield of hippocampus in ischemic brain
We have shown that the levels of PI 4-K, but not PI 3-K, in the CA1 subfield of the hippocampus and neuronal cells are reduced by brain ischemia and hypoxia, respectively. It is obvious that PI 4-K is a rate-limiting enzyme for PIP2 or PIP3 production. PIP2 is a well-known substrate for PI 3-K and is implicated in the subsequent cell-survival signaling. Although PIP2 represents less than 1% of total membrane phospholipids, it functions in a remarkable number of crucial cellular processes (Czech, 2000). For example, PIP2 is a precursor of intracellular second messengers such as diacylglycerol and inositol(1,4,5)P3 through its hydrolysis by phospholipase C. There have been some reports describing a relationship between brain ischemia and phospholipase C activation. Brain ischemia induces exclusively phospholipase C activation (Strosnajder, 1989), whereas levels of PI(4)P and PIP2 decrease rapidly at 30 minutes and then return transiently to the basal level at 1 to 6 hours after ischemia (Narita et al., 2000). Hence, it is suggested that the decrease in the PIP2 level observed here is dependent on the change in the PI 4-K level, but is not derived from the result of hydrolysis by phospholipase C. Besides this function, PIP2 has been shown to play an important role in cell-survival and cell-death signaling, in membrane trafficking (via endocytosis), and at the membrane/cytoskeletal interface via phagocytosis (Honda et al., 1999; Raucher et al., 2000; Toker, 1998). In particular, the association between PIP2 and gelsolin may be crucial when considering cell survival or apoptosis under severe conditions like ischemia. Gelsolin was originally identified as an actin-regulatory protein that promotes actin disassembly (Sun et al., 1999) and thereafter found to be a substrate for caspase-3, and the cleaved product accelerates apoptosis (Kothakota et al., 1997). Interestingly, there are two reports that gelsolin together with PIP2 can inhibit apoptosis (Azuma et al., 2000; Koya et al., 2000). As a possible mechanism, Azuma et al. (2000) proposed that complex of gelsolin and PIP2 selectively inhibits caspase-3 and caspase-9 related to apoptotic progression. However, Koya et al. (2000) showed that gelsolin together with PIP2 suppresses the loss of mitochondrial membrane potential and after cytochrome c release, though the mechanism has not been demonstrated yet. Thus, although the mode of action of gelsolin with PIP2 in preventing apoptosis is controversial, the potent inhibitory effect of this complex on apoptosis is evident. The findings presented here indicate that the decrease in PIP2 levels based on PI 4-K down-regulation was detected before specific delayed cell death in the CA1 area. Dissolution of the gelsolin–PIP2 complex linked to inhibition of apoptosis by brain ischemia may induce the delayed neuronal cell death in vivo.
In summary, we have demonstrated here that brain ischemia brings about the down-regulation of PI 4-K, but not PI 3-K, in the CA1 subfield of the hippocampus in rats. Furthermore, this decrease is related to reduced PIP2 levels in this area. Although the mechanism of the down-regulation of PI 4-K is not yet clear, it is of interest to address the crucial function of decreased PI 4-K or PIP2 levels in delayed neuronal cell death. PIP2 may play a role as a substrate or mediator of the cell-survival signal via the PI 3-K/Akt pathway, and may work in concert with gelsolin to prevent the activation of caspase or loss of mitochondrial membrane potential linked to apoptosis. Thus, to elucidate the detailed mechanism behind these findings may be therapeutically important, because the decrease in PI 4-K or PIP2 occurs before the characterized delayed loss of neurons in the hippocampus 3 to 5 days after transient forebrain ischemia, suggesting that the prevention/supplementation of PI 4-K or PIP2 losses during this period would provide a therapeutic effect after ischemic injury.
Footnotes
Abbreviations used:
Acknowledgments
The authors thank Dr. Sachiko Saino-Saito (Yamagata University) and Prof. Hisatake Kondo (Tohoku University) for providing us with rat PI 4-K cDNA.