
Abstract
Thrombolytic stroke therapy with tissue plasminogen activator (tPA) is limited by serious risks of intracerebral hemorrhage. In this study, the authors show that a novel antiactin-targeted immunoliposome significantly reduced tPA-induced hemorrhage in an established rat model of embolic focal stroke. Spontaneously hypertensive rats were subjected to focal ischemia using homologous blood clot emboli. Delayed administration of tPA (10 mg/kg, 6 hours after ischemia) induced intracerebral hemorrhage at 24 hours. In control rats treated with tPA plus vehicle, hemorrhage volumes were 9.0 ± 2.4 uL (n = 7). In rats treated with tPA plus antiactin immunoliposomes, hemorrhage volumes were significantly reduced to 4.8 ± 2.7 uL (n = 8, P < 0.05). No significant effects were seen when rats were treated with tPA plus a nontargeted liposome (7.8 ± 2.1 uL, n = 9). Fluorescent immunohistochemistry showed that rhodamine-labeled targeted liposomes colocalized with vascular structures in ischemic brain that stained positive for endothelial barrier antigen, a marker of cerebral endothelial cells. These data suggest that immunoliposomes may ameliorate vascular membrane damage and reduce hemorrhagic transformation after thrombolytic therapy in cerebral ischemia.
Thrombolytic therapy with tissue type plasminogen activator (tPA), when administered within 3 hours of onset, is effective for acute ischemic stroke (ECASS Study Group, 1995; NINDS rt-PA Stroke Study Group, 1995). However, treatment efficacy is accompanied with an elevated risk of intracerebral hemorrhage (Larrue et al., 1999; NINDS rt-PA Stroke Study Group, 1997). Hemorrhage is related in part to ischemic severity, but the precise mechanisms involved in vascular disruption have not been fully elucidated.
Animal models that allow for embolic arterial occlusions with homologous blood clots have recently been developed (Hara et al., 2000; Jiang et al., 2000; Lapchak et al., 2001; Overgaard, 1994; Shuaib et al., 2002). Delayed treatment with tPA has been observed to result in hemorrhagic transformations in these models (Brinker et al., 1999; Chopp et al., 1999; Kano et al., 2000; Lapchak et al., 2001). Oxidative damage is implicated because cotreatment with free-radical scavengers significantly reduces tPA-induced hemorrhage (Asahi et al., 2000; Lapchak et al., 2001). Because oxidative injury is likely to involve damage to vascular cell membranes, we hypothesized that a treatment that could “reseal” membranes may be useful. Such an approach uses targeted immunoliposomes that recognize intracellular antigens that become exposed in cells with damaged membranes. The targeted immunoliposomes would then selectively bind to these cells and merge with the damaged membranes. The validity of this idea has been shown in hypoxic myocytes in vitro (Khaw et al., 1995) and in an in vivo model of cardiac ischemia (Torchilin et al., 1996). Here, we test this immunoliposome approach in our model of tPA-induced hemorrhage in rat focal embolic ischemia.
MATERIALS AND METHODS
Rat model of embolic focal cerebral ischemia
All experiments were performed following an institutionally approved protocol in accordance with the National Institute of Health's Guide for the Care and Use of Laboratory Animals. Male spontaneously hypertensive (SH) rats (Taconic, Germantown, NY, U.S.A.) were used for all experiments. Animals were anesthetized with halothane (1% to 1.2%) under spontaneous respiration in a nitrous oxide/oxygen mixture. Rectal temperatures were maintained between 37°C and 38°C with a thermostat-controlled heating pad. The right femoral artery was cannulated, and physiologic parameters including rectal temperature, mean arterial blood pressure, pH, Pco2, and Po2 were monitored. The right femoral vein was cannulated for drug administration. Homologous blood clots were made using methods adapted and modified from Zhang et al. (1997). Briefly, femoral arterial blood from a donor rat was withdrawn into 50 cm of PE-50 tubing and kept in the tube for 2 hours at room temperature, and subsequently retained for 22 hours at 4°C. Five centimeters of the PE-50 tubing containing the clot was cut and connected to a syringe filled with saline using a 23-gauge needle. The clot was transferred into a dish filled with saline and washed. Then the clot was shifted to a modified PE-50 catheter with a 0.3-mm outer diameter filled with saline. Under a surgical microscope (Carl Zeiss, Inc., Thornwood, NY, U.S.A.), a modified PE-50 catheter with a 50-mm-long blood clot was gently inserted into the external carotid artery until the tip was positioned just proximal to the origin of middle cerebral artery. Then the clot in the catheter was injected into the internal carotid artery along with small amount of saline. After 5 minutes, the catheter was withdrawn from the external carotid artery, and rats were allowed to recover. At 6 hours after ischemic onset, rats were reanesthetized and reperfusion was achieved using tPA (10 mg/kg in 1 mL saline intravenously infused over a 20-minute period; Activase, Genentech Inc., San Francisco, CA, U.S.A.) at 6 hours after ischemia. The relatively high dose of tPA was chosen based on the approximately 10-fold difference in fibrin-specific enzyme activity between human and rodent systems (Korninger and Collen, 1981). Confirmation of ischemia and reperfusion was achieved using laser Doppler flowmetry. Laser Doppler flowprobes (Perimed) were stereotactically placed through a skull burr-hole at the center of the ischemic zone (2 mm posterior and 6 mm lateral to bregma). This follows previously established protocols that have been shown to effectively induce clot lysis and cerebral reperfusion in rat embolic stroke (Brinker et al., 1999; Chopp et al., 1999; Kano et al., 2000; Meng et al., 1999; Sumii and Lo, 2002). In rats treated with liposomes, an intraarterial pretreatment protocol was used in the present proof-of-concept study. The same catheters containing the clots were used. Each catheter was loaded with 3 mg of liposomes in a 30-uL volume (see Immunoliposome preparation for details). Catheters were inserted into the internal carotid artery as described previously, liposomes were gently infused, and clot injection immediately followed to achieve focal ischemia.
Analysis of infarct volumes and neurologic deficits
At 24 hours after ischemia, rats were assessed with a four-point neurologic deficit scale that has been extensively used in rat models of stroke (Bederson et al., 1986). After neurologic assessment, rats were killed with a lethal overdose of sodium pentobarbital and transcardially perfused to remove all intravascular blood. Coronal brain sections (2-mm thick) were stained with 2.3.5-triphenyltetrazolium chloride (TTC, Sigma, St. Louis, MO, U.S.A.). Infarct volumes were quantified using a computer-assisted image analysis techniques where TTC lesion areas from each slice were integrated to yield total ischemic lesion volumes (Aoki et al., 2002; Sumii and Lo, 2002). The indirect method was used to normalize for hemispheric swelling (Lin et al., 1993).
Spectrophotometric assay of intracerebral hemorrhage
Cerebral hemorrhage was quantified with a previously described spectrophotometric assay (Choudri et al., 1997) with some modification (Asahi et al., 2000; Sumii and Lo, 2002). Initially, a standard curve was obtained using a “virtual” model of hemorrhage. Hemispheric brain tissue was obtained from naive rats subjected to complete transcardial perfusion to remove intravascular blood. Incremental volumes of homologous blood (0, 2, 4, 8, 16, and 32 uL) were added to each hemispheric sample with PBS to reach a total volume of 3 mL, followed by homogenization for 30 seconds, sonication on ice for 1 minute, and centrifugation at 13,000 rpm for 30 minutes. Drabkin's reagent (1.6 mL, Sigma) was added to 0.4-mL aliquots and allowed to stand for 15 minutes at room temperature. Optical density was measured and recorded at 540 nm with a spectrophotometer (Spectronix 3000, Milton-Roy, Rochester, NY, U.S.A.). These procedures yielded a linear relationship between hemoglobin concentrations in perfused brain and the volume of added blood, as previously described (Asahi et al., 2000; Sumii and Lo, 2002). The reproducibility of this method was assessed by repeating spectrophotometric measurements on defined volumes of blood in test tubes (0.1, 1.0, 2.0, 4.0, and 8.0 uL). All measurements (n = 4 per group) yielded coefficients of variation (SD/mean) of 0.5% to 1.2%. Measurements from perfused brains subjected to ischemia and/or tPA reperfusion were compared with this standard curve to obtain data in terms of hemorrhage volume (uL). Hemorrhage measurements were performed on brains already stained with TTC for infarct quantitation. Pilot studies showed that TTC staining did not alter the spectrophotometric hemoglobin assay.
Immunoliposome preparation
Egg phosphatidylcholine, cholesterol, and the amphiphilic fluorescent dye rhodamine-phosphatidyl ethanolamine were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL, U.S.A.). All liposomes were prepared by detergent depletion technology using dialysis as method of detergent removal (Lasch et al., 2002). Lipids (phosphatidylcholine/cholesterol/rhodamine-phosphatidyl ethanolamine, 7:3:0.1 molar ratio) were solubilized in chloroform and transferred into a round-bottom flask. After the complete removal of the organic solvent using a rotary evaporator, the lipid film was resolubilized in Hank's balanced salt solution (HBS) containing 0.016-mol/L octylglycoside. Liposomes were formed by dialysis overnight against HBS at 4°C using membrane tubing with a molecular weight cut-off (MWCO) of 12 to 14,000 (Spectrum Laboratories Inc., Rancho Dominguez, CA, U.S.A.). For sizing, liposomes were passed through polycarbonate filters with the appropriate pore size (Nucleopore, 200 nm). For preparing antibody-bearing liposomes (immunoliposomes), antiactin antibodies were hydrophobized with N-glutaryl-phosphatidylethanolamine (NGPE) (purchased from Avanti Polar Lipids) in the presence of detergent followed by incorporation of the modified antibody into dialysis liposomes (Mori and Huang, 1999; Weissig et al., 1986). Briefly, 0.3 mg NGPE was dried with argon from a chloroform solution and solubilized with 0.5 mL 0.016-mol/L octylglycoside in 50-mmol/L 2-(4-morpholino)-ethane sulfonic acid (MES). The solution was supplemented with 12 mg water-soluble carbodiimide and 15 mg N-hydroxy-sulfosuccinimide. The resulting mixture was incubated for 5 minutes and then added to the solution of 2 mg of antibody in 0.1-mol/L HEPES (pH 7.6). The pH was adjusted immediately to 8.0 with 1-mol/L NaOH, and the mixture incubated overnight at 4°C. The modified antibody was then purified by dialysis against HBS and the appropriate amount was added into the octylglycoside solution of the lipid mixture prior to liposome formation via dialysis as described previously. This procedure results in the coupling of several hundred antibody molecules per single liposome with a size of about 200 to 250 nm (Torchilin et al., 2002).
Fluorescent immunohistochemistry
The colocalization of immunoliposomes with vascular structures was assessed in two rats at 3 hours after ischemic onset. After complete perfusion with phosphate-buffered saline (PBS), the brains were rapidly removed and frozen in liquid nitrogen vapor. Twenty-micrometer sections were cut on a cryostat (HM505E; Microm, Walldrof, Germany) and thaw-mounted onto precleaned glass slides and kept at −80°C until use. Thawed sections were dried completely and postfixed in absolute ethanol at −20°C for 10 minutes. After several washes in PBS, the sections were incubated with 10% normal goat serum (NGS) containing 0.3% Triton X-100 for 1 hour at 25°C. Then the sections were incubated with primary mouse antirat endothelial barrier antigen antibody (1: 3000; Sternberger Monoclonals Incorporated; Lutherville, MD, U.S.A.) diluted in 2% NGS, 0.3% Triton X-100, and 0.1% NaN3 in PBS for 12 hours at 4°C. After three rinses in PBS, sections were incubated with anti–mouse immunoglobulin M conjugated with Cy2 (1: 200; Jackson ImmunoResearch Laboratories; West Grove, PA, U.S.A.) for 1 hour at 25°C. Immunoliposomes that incorporated rhodamine were used, and colocalization with Cy2 (fluorescein isothiocyanate-conjugated)-labeled vascular endothelial cells was confirmed with a fluorescent microscope coupled to a digital imaging system (Olympus BX51; Tokyo, Japan) (Mori et al., 2002).
Statistical analysis
The major quantitative endpoints used in all experiments were infarct volume and calculated hemorrhage volume. Data are expressed as mean ± SD. Distributions were checked with Shapiro-Wilks tests and found to be normal, so parametric tests were used. Analysis of variance was used for the multiple-group comparisons, followed by Tukey HSD tests for intergroup comparisons.
RESULTS
Systemic parameters were within normal range during experimental procedures. Laser Doppler flowmetry showed reduction of perfusion to below 20% of baseline after clot injection in all rats, confirming that focal cerebral ischemia was successfully induced. In all rats, tPA was infused 6 hours after ischemic onset. Twenty-four hours later, hemoglobin spectrophotometry showed that cerebral hemorrhage volumes were significantly reduced in rats treated with antiactin immunoliposomes (4.8 ± 2.7 uL) compared with rats treated with plain liposomes (7.8 ± 2.1 uL) or vehicle (9.0 ± 2.4 uL) (Fig. 1A). In contrast, ischemic lesion volumes quantified by tetrazolium staining showed that there were no significant differences among rats treated with vehicle, plain liposomes, or targeted immunoliposomes (Fig. 1B).
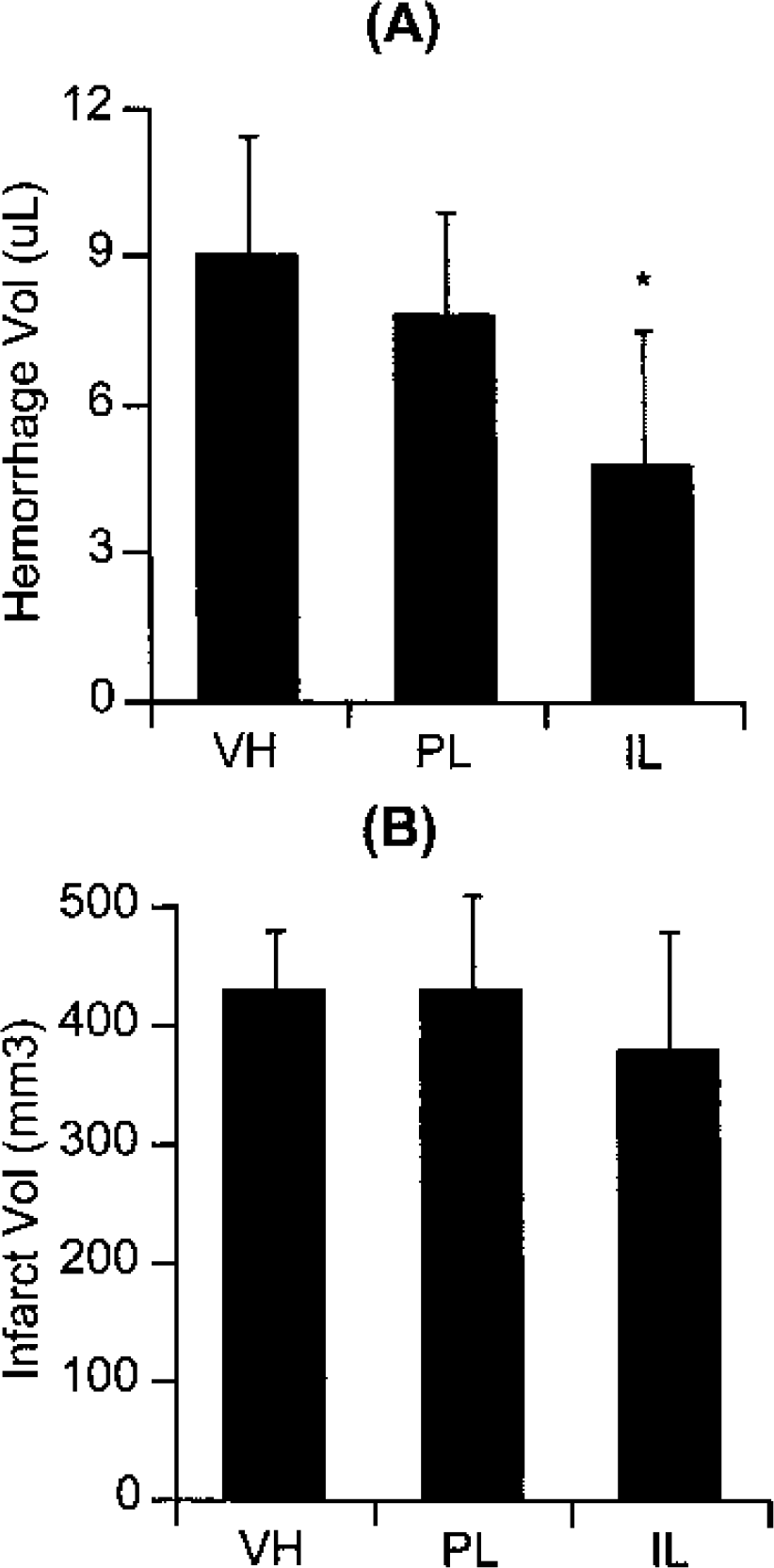
Effects of liposomes on tissue plasminogen activator (tPA)-induced cerebral hemorrhage in rat embolic (clot-based) focal ischemia.
To confirm the vascular distribution of the liposomes in ischemic brain, targeted immunoliposomes fluorescently labeled with rhodamine were used. Immunohistochemistry showed that antibodies against EBA, an antigen specific for cerebral endothelial cells in rat brain (Lin and Ginsberg, 2000), clearly stained cerebrovasculature in all brains. Rhodamine-labeled immunoliposomes demonstrated significant colocalization with EBA-stained vessels (Fig. 2A and Fig. 2B).
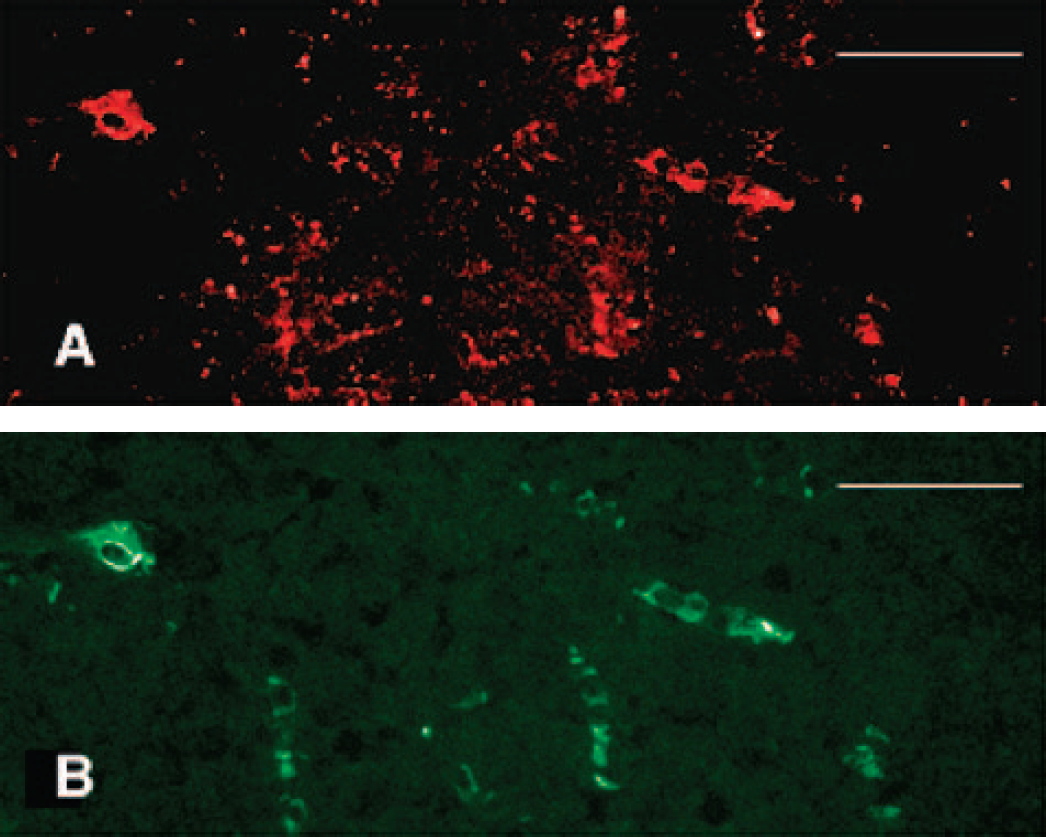
Immunohistochemistry showing the vascular colocalization of antiactin immunoliposomes in ischemic cortex 3 hours after ischemic onset.
DISCUSSION
Thrombolysis with tPA can increase risks of cerebral hemorrhage after ischemic stroke (Larrue et al., 1999; NINDS rt-PA Stroke Study Group, 1997). Thus, it is important to dissect the mechanisms involved and to identify methods that can ameliorate hemorrhage risks after tPA stroke therapy. Reperfusion injury after ischemia involves mechanisms of oxidative stress and injury (Akins et al., 1996; Chan, 1996; Hallenback and Dutka, 1990). Oxidative injury may play a role in mediating cerebrovascular damage after thrombolytic stroke therapy, thus predisposing brain tissue to hemorrhage (del Zoppo et al., 1986; Lyden and Zivin, 1993). Accumulating evidence suggests that free-radical damage to membrane compartments of cerebral blood vessels underlies this phenomenon of tPA-induced hemorrhagic transformation (Asahi et al., 2000; Lapchak et al., 2001). We have previously shown that immunoliposomes can target and reseal damaged cellular membranes (Khaw et al., 1995). In the present study, we show that this novel approach can ameliorate tPA-induced vascular injury and subsequent hemorrhage in a rat model of thromboembolic focal ischemia.
The rationale behind this study is centered on the idea that lipids from liposomes can reseal damaged cellular membranes. There is an accumulating literature suggesting that after oxidative or mechanical injury to cells, membrane disruption plays a central role in mediating ionic imbalance and subsequent cell dysfunction and death (Borgens, 2001). Hence, any method that can reestablish cell membrane integrity should theoretically be able to help restore cellular function. In animal models of spinal cord injury, treatment with the hydrophilic polymer polyethylene glycol significantly repaired axonal and somal membranes and promoted recovery of action-potential conduction as well as whole-animal behavioral function (Borgens and Shi, 2000; Luo et al., 2002). Because it is well known that liposomes can fuse with cell membranes, the present study used immunoliposomes to target vascular cells with exposed intracellular actin cytoskeleton, and thereby “patch” holes in the membrane induced by ischemia-reperfusion injury. Insofar as hemorrhagic transformation may involve damaged membranes in the cerebrovasculature, the immunoliposomes used here should reseal damaged membranes and ameliorate vascular leakage.
Many questions remain. First, the specific molecular mechanisms should be elucidated in more detail. Although it has been shown that a significant part of immunoliposomes fuses with damaged cell membranes (Khaw et al., 2001), it is not completely clear if this is the primary mechanism responsible for restoration of membrane integrity in damaged cells or whether a simple “plugging” of membrane holes with the entire immunoliposome can also play a role. A second caveat involves the specificity of the events that occur. Although we show that fluorescently labeled immunoliposomes colocalize with brain endothelial antigens, there was also signal from nonvascular compartments. Other damaged cells (e.g., neurons, astrocytes) would also have damaged membranes that can attract immunoliposomes. In fact, our choice of antiactin as the targeting moiety was intentionally nonspecific. Whatever damaged membranes one can reseal would be beneficial in our model. This idea is consistent with the finding that nontargeted plain liposomes had no effect on hemorrhage. Future studies that more carefully assess cellular distributions are needed to confirm this finding. Finally, it is interesting to note that although immunoliposomes decreased hemorrhage, there was no effect on ischemic infarction as measured by tetrazolium staining. This is somewhat surprising because previous studies had shown that antimyosin immunoliposomes reduced myocardial infarction in vivo (Khaw et al., 2000) and decreased cytotoxicity in cultured myocytes exposed to hypoxia in vitro (Khaw et al., 1995). Possible reasons for the lack of efficacy here may include the severity of damage, differences in liposome penetration into parenchyma, and overall differences in brain versus heart ischemic pathology. Nevertheless, the equivalent infarcts at least assure us that similar insults were present, and the reduction in hemorrhage severity was not simply due to differences in ischemia per se. The relationships between infarction, hemorrhage, and neurologic outcomes (not measured in the present study) will also have to be carefully assessed. Even if there are no effects on infarct size, it remains possible that amelioration of cerebral hemorrhagic transformation may be beneficial.
In conclusion, our findings suggest that anticytoskeletal immunoliposomes may be used for ameliorating vascular damage and reducing hemorrhage associated with thrombolytic stroke therapy. Clearly, the underlying molecular mechanisms involved remain to be fully established. However, these data provide proof of principle for therapies focused on cerebrovascular damage presumably caused by reperfusion injury. The emerging concept of the neurovascular unit in stroke research emphasizes that, although neuronal cell death ultimately underlies ischemic brain damage, endothelial dysfunction is a proximal trigger for parenchymal injury (Lo et al., 2003). Perhaps even more importantly, the present data suggest that immunoliposome strategies may offer unique methods for targeting the cerebral endothelium in stroke. Additional studies using this model are warranted to explore the mechanisms involved, test therapeutic windows, and translate these findings into potential clinical applications.