
Abstract
Thrombolytic stroke therapy with tissue plasminogen activator (tPA) remains complicated by serious risks of cerebral hemorrhage and brain injury. In this study, a novel model of tPA-induced hemorrhage was used in spontaneously hypertensive rats to examine the correlates of hemorrhage, and test methods of reducing hemorrhage and brain injury. Homologous blood clot emboli were used to occlude the middle cerebral artery in spontaneously hypertensive rats, and delayed administration of tPA (6 hours postischemia) resulted in high rates of cerebral hemorrhage 24 hours later. Compared with untreated rats, tPA significantly increased hemorrhage volumes by almost 85%. Concomitantly, infarction and neurological deficits were worsened by tPA. A parallel experiment in normotensive Wistar-Kyoto rats showed markedly reduced rates of hemorrhage, and tPA did not significantly increase hemorrhage volumes. To examine whether tPA-induced hemorrhage was caused by the delayed onset of reperfusion per se, another group of spontaneously hypertensive rats was subjected to focal ischemia using a mechanical method of arterial occlusion. Delayed (6 hours) reperfusion via mechanical means did not induce hemorrhage. However, administration of tPA plus delayed mechanical reperfusion significantly increased hemorrhage volumes. Since reperfusion injury was implicated, a final experiment compared outcomes in spontaneously hypertensive rats treated with tPA plus the free radical spin trap α-phenyl tert butyl nitrone (α-PBN) versus tPA alone. tPA-induced hemorrhage volumes were reduced by 40% with α-PBN, and infarction and neurological deficits were also decreased. These results indicate that (1) blood pressure isanimportant correlate of tPA-induced hemorrhage, (2) tPA interacts negatively with reperfusion injury to promote hemorrhage, and (3) combination therapies with anti-free radical treatments may reduce the severity of tPA-induced hemorrhage and brain injury after cerebral ischemia.
Keywords
Although thrombolytic therapy may be effective for acute ischemic stroke (ECASS Study Group, 1995; NINDS rt-PA Stroke Study Group, 1995), there is an increased risk of cerebral hemorrhage (Larrue et al., 1999; NINDS rt-PA Stroke Study Group, 1997). In rodent models of embolic focal ischemia induced with homologous blood clots, delayed treatment with tissue plasminogen activator (tPA) has been observed to result in hemorrhagic transformations (Brinker et al., 1999; Chopp et al., 1999; Kano et al., 1999).
Here, we describe a quantitative model of tPA-induced hemorrhage in rats, and show that whereas blood pressure is an important correlate for hemorrhage, delayed onset of reperfusion per se is not. However, reperfusion injury appeared to interact negatively with tPA to promote hemorrhage. Importantly, combination treatment with the free radical spin trap α-phenyl tert butyl nitrone (α-PBN) significantly reduced tPA-induced cerebral hemorrhage and brain injury in embolic focal ischemia.
METHODS AND MATERIALS
Animal models of focal cerebral ischemia
All experiments were performed following an institutionally approved protocol in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Male Wistar Kyoto (WK) rats or spontaneously hypertensive (SH) rats (Taconic, Germantown, NY, U.S.A.) were used. Animals were anesthetized with halothane (1-1.2%) under spontaneous respiration in an air-oxygen mixture. Rectal temperatures were maintained between 37°C to 38°C with a thermostat-controlled heating pad. The right femoral artery was cannulated, and physiologic parameters including rectal temperature, mean arterial blood pressure, pH, P
For the embolic model, homologous blood clots were made using methods adapted and modified from Zhang et al (1997). Briefly, femoral arterial blood from a donor rat was withdrawn into 50 cm of PE-50 tubing and kept in the tube for 2 hours at room temperature, and subsequently retained for 22 hours at 4°C. Five centimeters of the PE-50 tubing containing the clot was cut and connected to a syringe filled with saline using a 23-gauge needle. The clot was transferred into a dish filled with saline and washed. Then the clot was shifted to a modified PE-50 catheter with a 0.3-mm outer diameter filled with saline. Under a surgical microscope (Carl Zeiss, Inc., Thornwood, NY, U.S.A.), a modified PE-50 catheter with a 50-mm long blood clot was gently inserted into the external carotid artery until the tip was positioned just proximal to the origin of middle cerebral artery. Then the clot in the catheter was injected into the internal carotid artery along with small amount of saline. After 5 minutes, the catheter was withdrawn from the external carotid artery. Reperfusion was achieved using tPA.
In the appropriate groups, human recombinant tPA (Activase, Genentech Inc, San Francisco, CA, U.S.A.) was administered intravenously (10 mg/kg in 1 mL of saline over 30 minutes) using previously established protocols that have been shown to effectively induce clot lysis and cerebral reperfusion in rat embolic stroke (Brinker et al., 1999; Chopp et al., 1999; Kano et al., 1999; Meng et al., 1999). The relatively high dose of tPA was chosen based on the approximately tenfold difference in fibrin-specific enzyme activity between human and rodent systems (Korninger and Collen, 1981).
For the mechanical model of ischemia and reperfusion, a standard intraluminal method was used (Lo et al., 1998; Zea Longa et al., 1989). Under a surgical microscope (Carl Zeiss), the right common, internal, and external carotid arteries were exposed, and a silicon-coated 4.0 nylon monofilament was inserted into the external carotid artery and advanced into the internal carotid artery until the tip occluded the proximal stem of the intracranial middle cerebral artery. Reperfusion was accomplished by removing the filament. To study the effects of tPA plus reperfusion, tPA was also administered in this mechanical model at the onset of filament withdrawal.
Analysis of infarct volumes and neurological deficits
At 24 hours after ischemia, rats were assessed with a 4-point neurological deficit scale that has been extensively used for rat models of stroke (Bederson et al., 1986). After neurological assessment, rats were killed with a lethal overdose of sodium pentobarbital and transcardially perfused to remove all intravascular blood. Coronal brain sections (2 mm thick) were stained with 2.3.5-triphenyltetrazolium chloride (TTC, Sigma, St. Louis, MO, U.S.A.). Infarct volumes were quantified using computer-assisted image analysis techniques where TTC lesion areas from each slice were integrated to yield total ischemic lesion volumes (Lo et al., 1998; Newcomb et al., 1998; Shimizu-Sasamata et al., 1998).
Spectrophotometric assay of intracerebral hemorrhage
Cerebral hemorrhage was quantified with a previously described spectrophotometric assay (Choudri et al., 1997) with some modification. Initially, a standard curve was obtained using a “virtual” model of hemorrhage. Hemispheric brain tissue was obtained from naive rats subjected to complete trans-cardial perfusion to remove intravascular blood. Incremental volumes of homologous blood (0, 2, 4, 8, 16, 32 μL) were added to each hemispheric sample with phosphate buffered saline (PBS) to reach a total volume of 3 mL, followed by homogenization for 30 seconds, sonication on ice for 1 minute, and centrifugation at 13, 000 rpm for 30 minutes. Drabkin's reagent (1.6 mL, Sigma) was added to 0.4 mL aliquots and allowed to stand for 15 minutes at room temperature. Optical density was measured and recorded at 540 nm with a spectrophotometer (Spectronix 3000, Milton-Roy, Rochester, NY, U.S.A). These procedures yielded a linear relationship between hemoglobin concentrations in perfused brain and the volume of added blood. Measurements from perfused brains subjected to ischemia and/or tPA reperfusion were compared with this standard curve to obtain data in terms of hemorrhage volume (μL). Hemorrhage measurements were performed on brains already stained with TTC for infarct quantitation. Pilot studies showed that TTC staining did not alter the spectrophotometric hemoglobin assay.
Statistical analysis
The major endpoints used in all experiments were infarct volume, hemorrhage volume, and neurological deficits. Data were expressed as mean ± SD. Distributions were checked with Shapiro-Wilks tests and found to be normal, so parametric tests were used. Analysis of variance was used for the multiple group comparisons, followed by Tukey's Honestly Significant Difference tests for inter-group comparisons. Correlations between infarct volume and hemorrhage severity were performed using linear regressions.
RESULTS
Experiment 1: tPA-induced hemorrhage and exacerbated brain injury in spontaneously hypertensive rats but not normotensive Wistar-Kyoto rats after embolic focal ischemia
Focal cerebral ischemia was induced using homologous clot emboli in four groups of rats to examine rates of tPA-induced hemorrhage and brain injury, and its relationship to blood pressure as a correlate: Group 1 (SH rats no treatment, n = 8), Group 2 (SH rats treated with tPA at 6 hours, n = 10), Group 3 (WK rats no treatment, n = 8), and Group 4 (WK rats treated with tPA at 6 hours, n = 9).
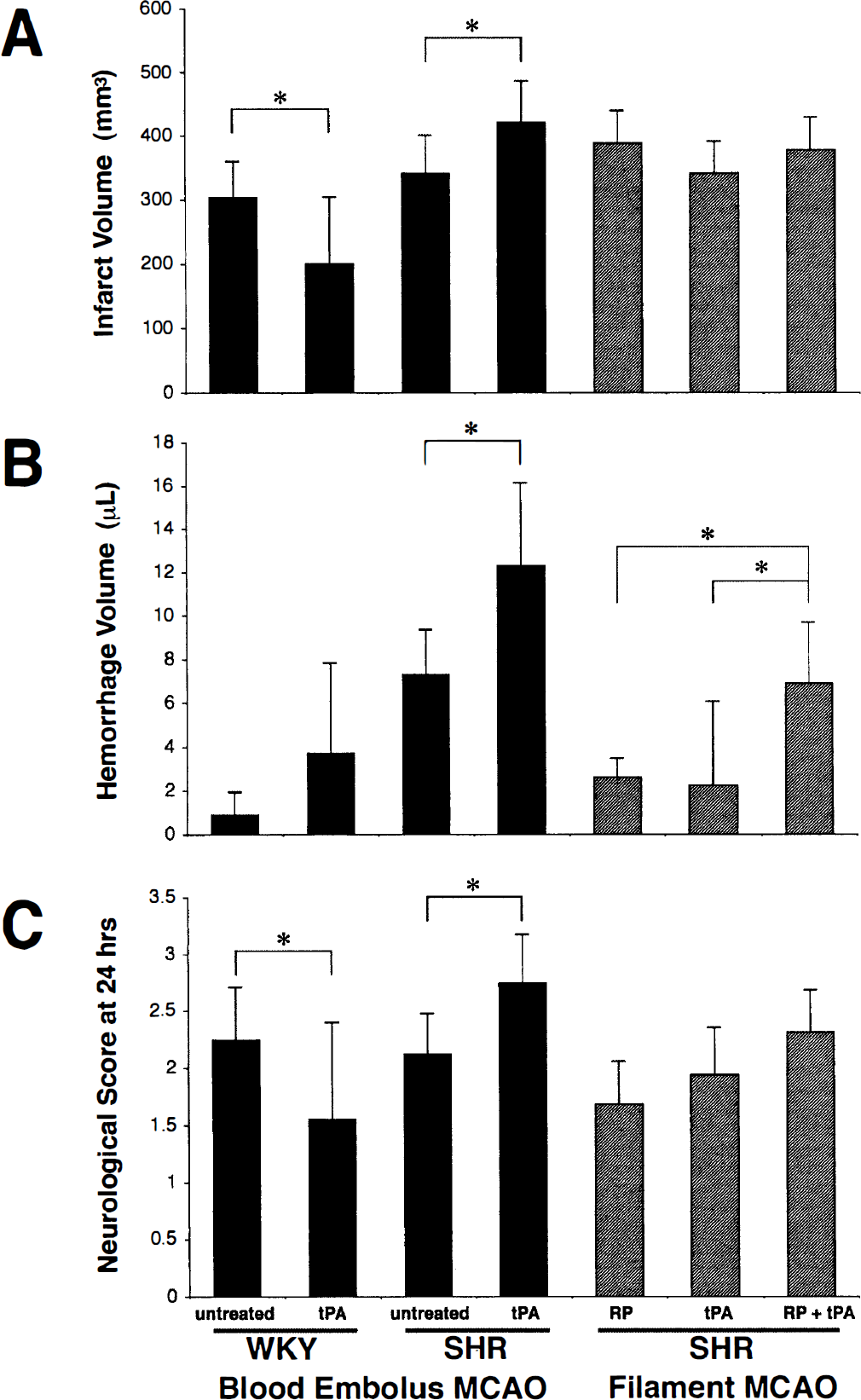
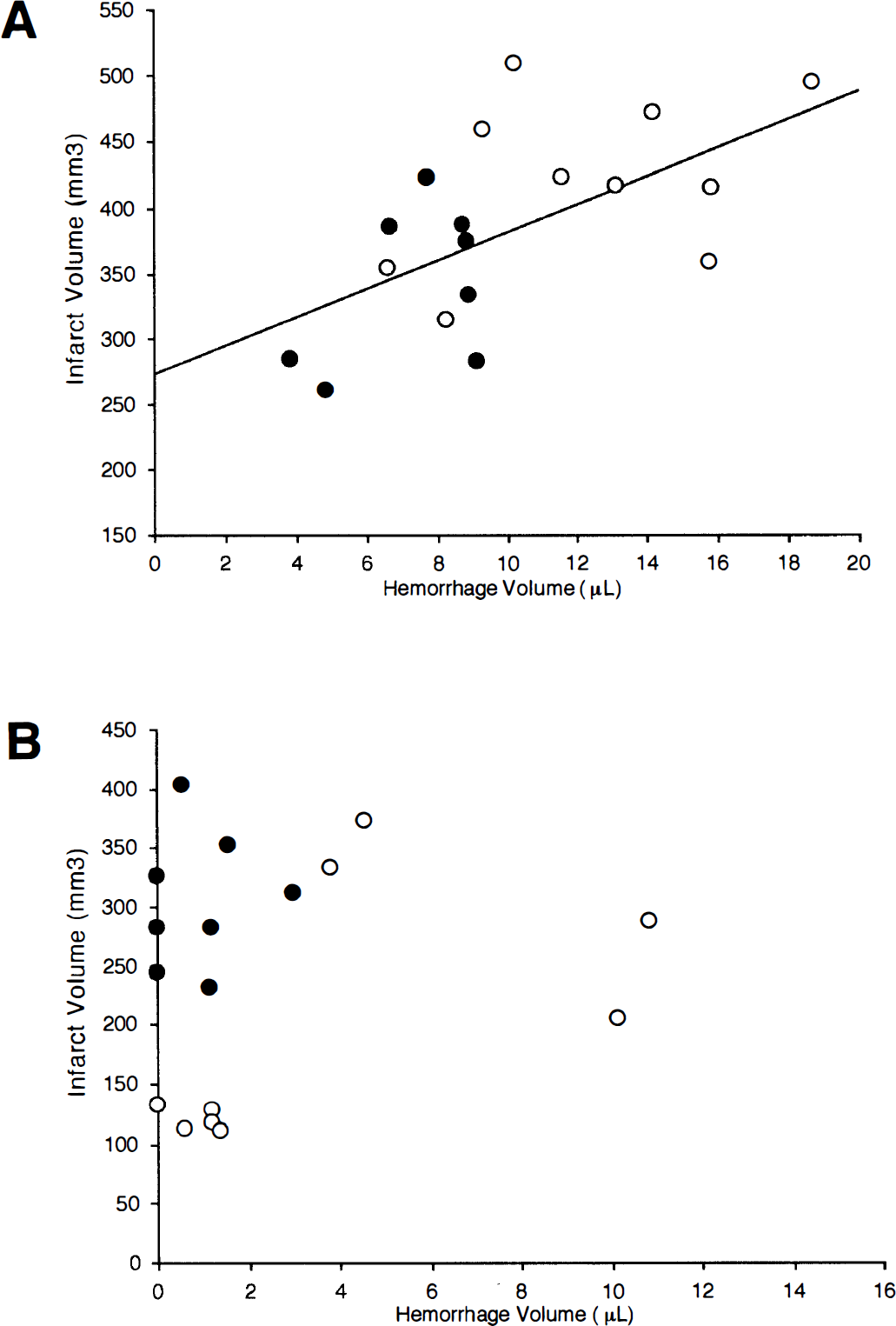
Systemic parameters remained within normal range for all rats, with SH rats showing clear hypertension (Table 1). Focal ischemic damage in the middle cerebral artery was present in all occluded rats after 24 hours. In SH rats, tPA induced hemorrhage in the middle cerebral artery territories (Fig. 1). Hemorrhage severity were significantly increased by tPA when compared with untreated rats (12.3 ± 3.8 mm3 in tPA-treated rats versus 7.3 ± 2.0 mm3 in untreated controls, P = 0.001, Fig. 2). Concomitantly, infarct volumes and neurological deficits were also worsened by tPA (Fig. 2). The severity of hemorrhage was positively correlated with infarct volumes (r = 0.611, P < 0.01, Fig. 3A). There was no correlation between hemorrhage and neurological deficits.
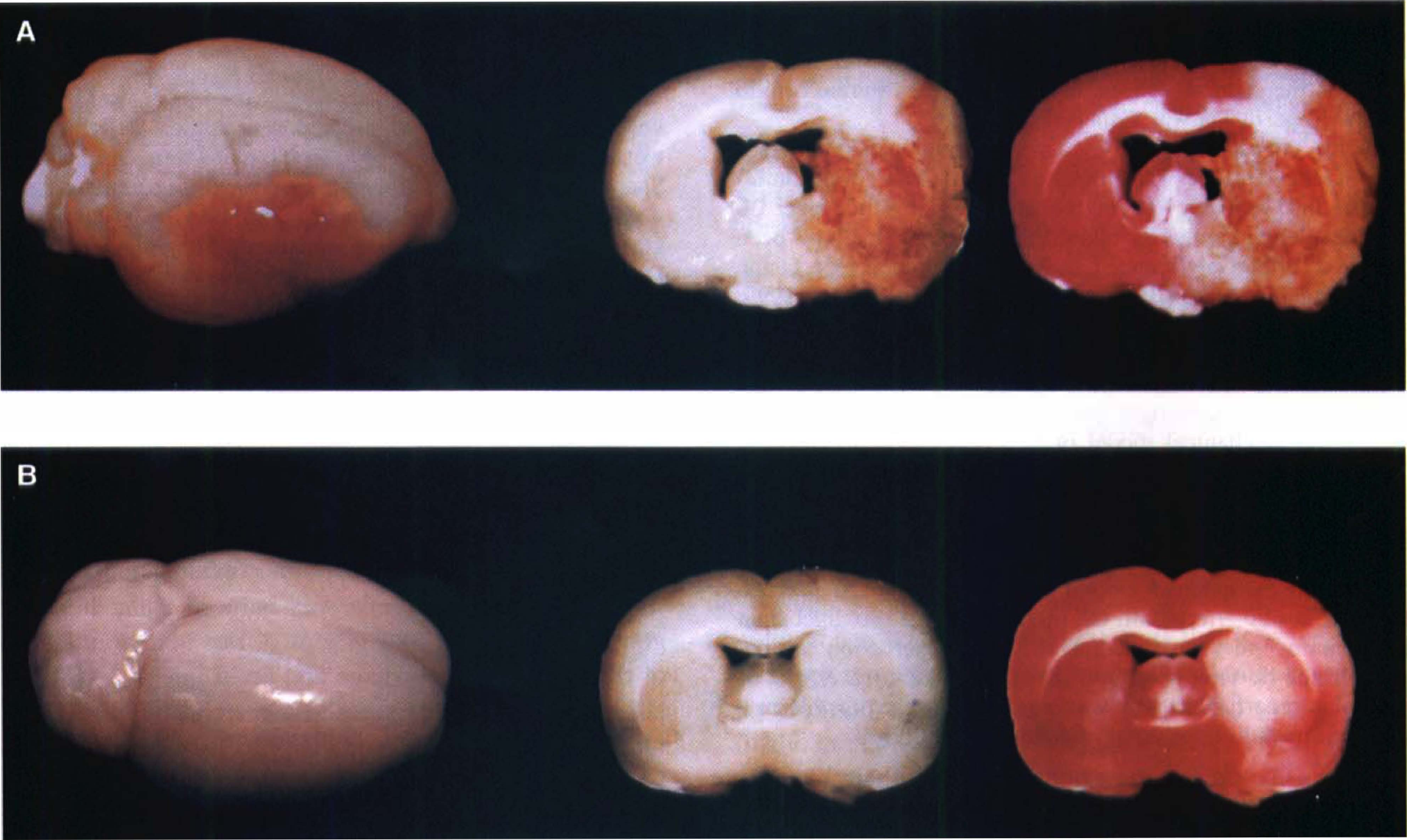
Hypertensive SH rats and normotensive Wistar Kyoto (WK) rats were subjected to embolic focal cerebral ischemia. At 6 hours after ischemic onset, rats were treated with tissue plasminogen activator (tPA). tPA induced extensive hemorrhagic transformation in SH rats (
(
Physiologic variables
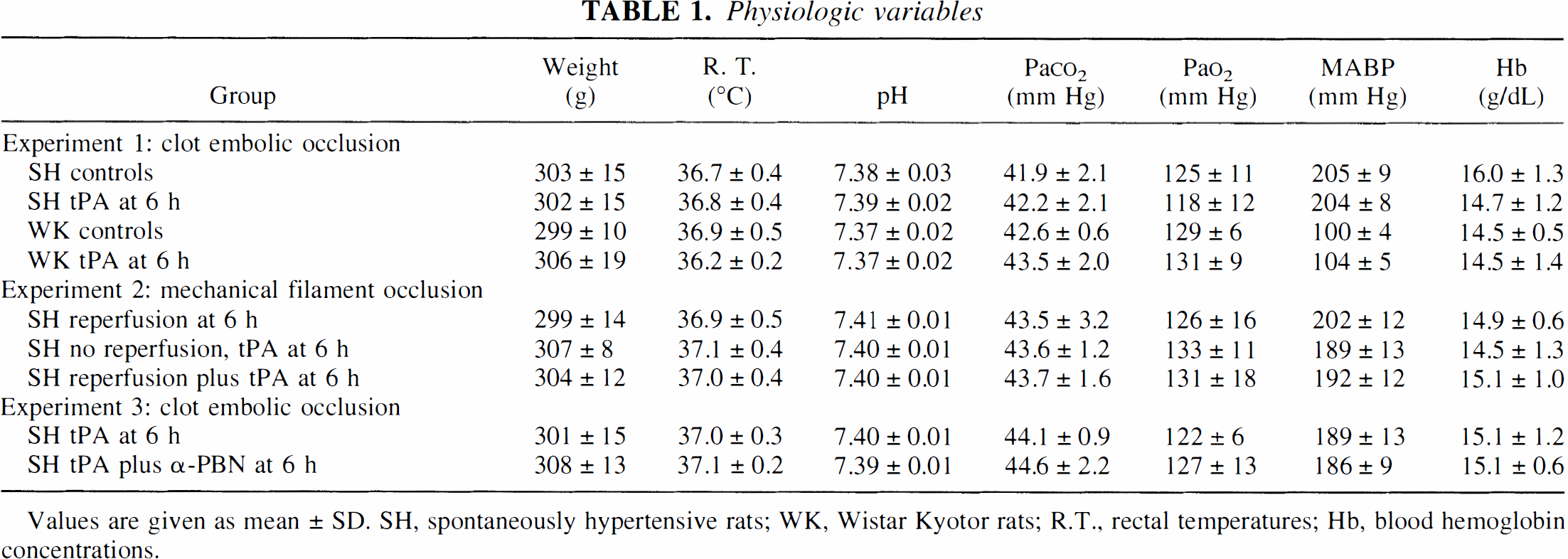
Values are given as mean ± SD. SH, spontaneously hypertensive rats; WK, Wistar Kyotor rats; R.T., rectal temperatures; Hb, blood hemoglobin concentrations.
Overall hemorrhage severity was much lower in normotensive WK rats (Fig. 1). Although hemorrhage severity appeared to be slightly increased by tPA, the difference did not reach statistical significance (Fig. 2). In contrast to the hypertensive SH rats, 24 hour infarct volumes and neurological deficits were significantly improved by tPA in normotensive WK rats (Fig. 2). There was no correlation between hemorrhage severity, infarct volumes, or neurological deficits (Fig. 3B).
(
Experiment 2: tPA interacted negatively with reperfusion injury to promote cerebral hemorrhage
The mechanical model of ischemia was used in three groups of rats to determine whether hemorrhage was induced by the onset of delayed reperfusion per se, tPA alone, or negative interactions between reperfusion injury and tPA: Group 1 (SH rats subjected to delayed mechanical reperfusion at 6 hours, n = 8), Group 2 (SH rats subjected to permanent focal ischemia and administered tPA at 6 hours, n = 8), and Group 3 (SH rats mechanically reperfused at 6 hours and administered tPA, n = 8).
Systemic parameters were normal in all rats. Twenty four hour infarct volumes were typical for this well-described mechanical model of focal cerebral ischemia (Fig. 2). Delayed reperfusion per se or administration of tPA in permanent focal ischemia did not induce high rates of hemorrhage (Fig. 2). However, cerebral hemorrhage was significantly elevated in rats exposed to both tPA plus delayed reperfusion (Fig. 2). Overall hemorrhage severity was lower in this series compared with the embolic clot series in experiment 1. There was no statistically detectable difference in infarct volumes or neurological deficits between all three groups (Fig. 2).
Experiment 3: Combination therapy with α-PBN reduced tPA-induced hemorrhage and brain injury
Since experiment 2 implicated a role for reperfusion injury in tPA-induced hemorrhage, we tested the effectiveness of the free radical spin trap α-PBN for reducing hemorrhage and brain injury in the embolic ischemia model: Group 1 (SH rats treated with tPA at 6 hours, n = 8) and Group 2 (SH rats treated with tPA plus 20 mg/kg α-PBN at 6 hours, n = 8).
All systemic parameters were within normal range (Table 1). Delayed tPA administration induced hemorrhage as expected (Fig. 4). Combination treatment with α-PBN significantly reduced hemorrhage severity (6.7 ± 4.2 mm3 in α-PBN plus tPA-treated rats versus 11.1 ± 3.8 mm3 in rats administered tPA alone, P = 0.004, Fig. 4). Concomitantly, infarct volumes and neurological outcomes were significantly improved as well (Fig. 4).
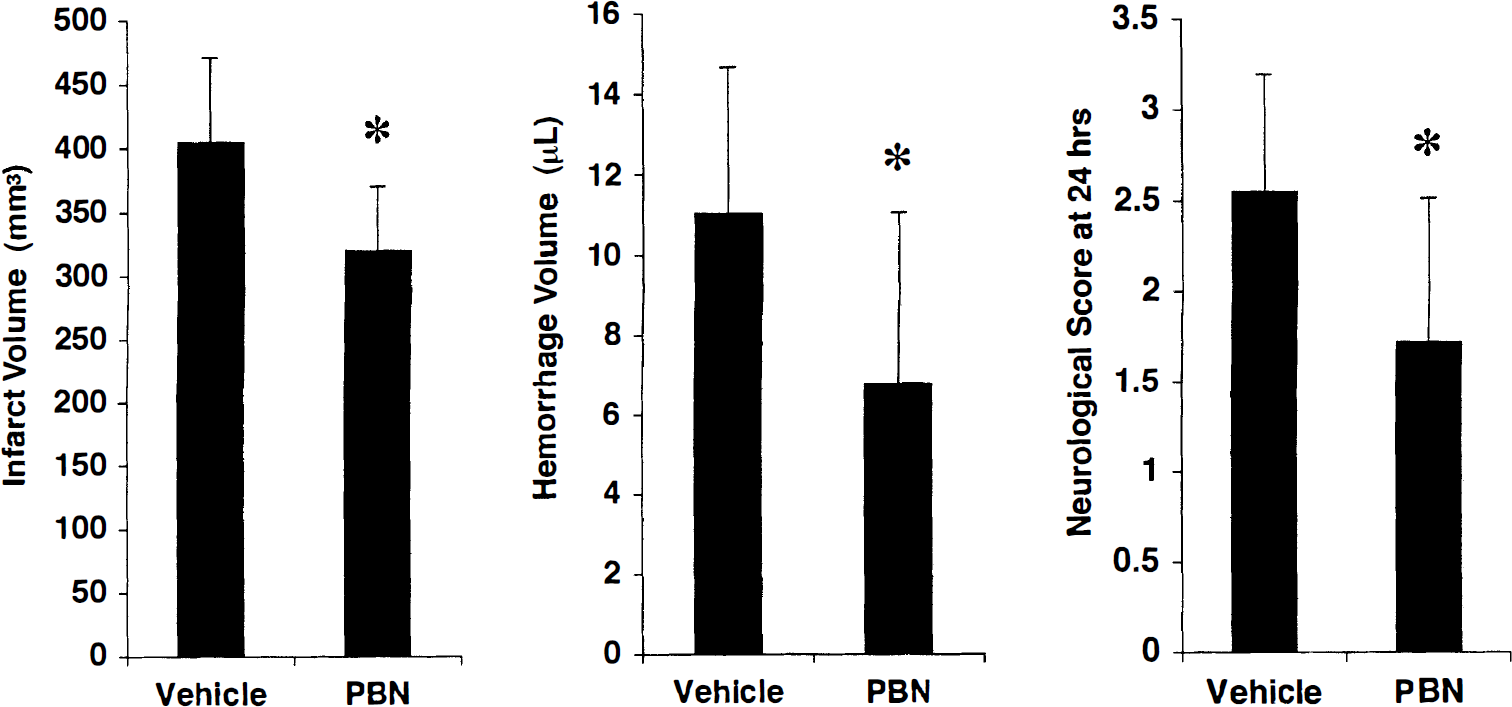
In hypertensive SH rats subjected to clot-induced embolic stroke, tissue plasminogen activator-associated hemorrhage was significantly reduced by cotreatment with the free radical spin trap α-phenyl tert butyl nitrone (PBN). Concomitantly, infarct volumes were reduced and neurological deficits were also improved. *P < 0.05.
DISCUSSION
Thrombolysis with tPA can increase risks of cerebral hemorrhage after ischemic stroke (Larrue et al., 1999; NINDS rt-PA Stroke Study Group, 1997). Thus, it is important to dissect the mechanisms involved, and identify methods that can ameliorate hemorrhage risks after tPA stroke therapy. In this report, we described a novel quantitative animal model of thrombolysis-induced hemorrhage in ischemic stroke. We showed that (1) blood pressure was a critical correlate because tPA induced hemorrhage in hypertensive SH rats but not normotensive WK rats, (2) delayed onset of reperfusion per se was not a major factor although tPA appeared to interact negatively with reperfusion injury to promote hemorrhage, and (3) combination treatment with the free radical spin trap α-PBN significantly reduced tPA-induced hemorrhage and brain injury after embolic cerebral ischemia.
Clinical data suggest that high systemic blood pressure may increase risks of cerebral hemorrhage (Levy et al., 1994). Here, parallel experiments were conducted in normotensive WK rats, which is the strain most closely related to the hypertensive SH rats. Overall, hemorrhage severity was much lower in normotensive WK rats compared to the SH rats, and tPA did not significantly increase cerebral hemorrhage. Although we cannot unequivocally exclude a role for other genetic differences in the hypertensive SH rats, it is likely that increased blood pressure may be the most parsimonious explanation here. These data can be interpreted as a validation of our model since the positive correlation between hemorrhage and blood pressure was to be expected.
The high rates of hemorrhage in our model may have been caused by the delayed onset of tPA treatment (6 hours postocclusion) rather than being related to tPA thrombolysis per se. To exclude this potential confound, studies were conducted in the standard filament model of arterial occlusion with a delayed onset of mechanical reperfusion at 6 hours. No significant hemorrhage was induced, suggesting that tPA-induced hemorrhage was not solely caused by delayed reperfusion injury. However, when tPA was added to the mechanical reperfusion model, hemorrhage rates were significantly elevated. These data suggest that tPA may interact negatively with processes of reperfusion injury, thus leading to damaged cerebrovasculature and hemorrhage. tPA has been shown to amplify excitotoxic and ischemic neuronal injury (Tsirka et al., 1996, 1997; Wang et al., 1998). Recently, it was also demonstrated that tPA amplified hemoglobin-induced toxicity in neuronal cultures (Wang et al., 1999). Similar, albeit undefined, cytotoxic effects of tPA may be involved here.
Reperfusion injury after ischemia involves mechanisms of oxidative stress and injury (Akins et al., 1996; Chan, 1996; Hallenback and Dutka, 1990). Oxidative injury may play a role in mediating cerebrovascular damage after thrombolytic stroke therapy, thus predisposing brain tissue to hemorrhage (del Zoppo et al., 1986; Lyden and Zivin, 1993). An important finding in the present study was the reduction of tPA-induced hemorrhage and brain injury by the free radical spin trap α-PBN. These findings suggest that compounds targeted against oxidative injury may prove valuable when used as combination treatments for ameliorating hemorrhage risks associated with thrombolytic stroke therapy. Clearly, the underlying molecular mechanisms involved remain to be fully established. However, these data show that tPA-induced hemorrhage can be reduced by combination therapies. Additional studies using this model are warranted to explore the mechanisms involved, test therapeutic windows, and translate these findings into clinical applications.
Footnotes
Acknowledgments
The authors express their gratitude to Dr. Michael Chopp for sharing his methodology for rodent embolic focal ischemia models. The authors also thank Drs. Michael Moskowitz, Bruce Rosen, and Rick Dijkhuizen for their generous intellectual support and invaluable discussions.