
Abstract
Inflammation has been widely perceived as participating in the etiology of acute and chronic neurodegenerative conditions. Accordingly, in the context of traumatic injuries or chronic neurodegenerative diseases in the central nervous system (CNS), activated microglia have been viewed as detrimental and attempts have been made to treat both conditions by antiinflammatory therapy. Recent studies have suggested that microglia act as stand-by cells in the service of both the immune and the nervous systems. In the healthy CNS these cells are quiescent, but in the event of injury to axons or cell bodies they exercise their neural function by buffering harmful self-compounds and clearing debris from the damaged site, and their immune function by providing immune-related requirements for recovery. Proper regulation of the inflammatory (autoimmune) response to injury will arrest degeneration and promote regrowth, whereas inappropriate regulation will lead to ongoing degeneration. Regulation is achieved by the operation of a T cell–mediated response directed to abundant self-antigens residing in the damaged site. Since this immune-dependent mechanism was found to protect against glutamate toxicity (a major factor in neurodegenerative disorders), boosting of this response might constitute the basis for development of a therapeutic vaccination against neurodegenerative diseases, all of which exhibit similar pathways and patterns of progression.
THE CENTRAL NERVOUS SYSTEM AS AN IMMUNE-PRIVILEGED SITE
The unique interaction between the central nervous system (CNS) and the immune system has long been regarded as a relation characterized by immune privilege, in which local immune responses within the CNS are restricted (Streilein, 1993, 1995). Unlike most peripheral tissues, the CNS functions through a network of postmitotic cells (neurons) that are incapable of regeneration and hence cannot be replaced when aging or impaired. Being simultaneously essential for survival and unable to recover fully from injury, the CNS is in special need of protection from pathogens and insults. However, the CNS is also vulnerable to damage that might be caused by the very means that the immune system uses to defend peripheral tissues from pathogens. Consequently, immune privilege in the CNS has been viewed as an evolutionary adaptation developed to protect the intricate neuronal networks of the CNS from incursion by the immune system (Lotan and Schwartz, 1994).
An early definition of CNS immune privilege was based on the assumption that the immune system ignores the CNS. This assumption was supported by the observed tendency of the CNS not to reject allografts (tissue grafts from the same species but from a different major histocompatibility complex [MHC] haplotype) (Medawar, 1948). Immune privilege was thought to be maintained by the isolation of antigens within the CNS and the inability of immune cells to enter the CNS under normal physiologic conditions. Any leukocyte entry was viewed as evidence of pathology. Several observations have challenged this definition. First, CNS antigens can escape and induce immune responses in the host (Cserr et al., 1992; Cserr and Knopf, 1992). Second, activated T cells have been found to enter the CNS in the absence of discernible neuropathology (Flugel et al., 2001; Hickey et al., 1991; Shrikant and Benveniste, 1996). Third, leukocyte recruitment into the CNS appears to successfully resolve some CNS viral infections, such as Sindbis virus encephalitis, without the development of any apparent long-term bystander effects (Griffin et al., 1997). Fourth, based on the relatively prolonged survival of xenografts (tissue grafts from different species) in immunosup-pressed persons, it was suggested that the immune system participates in CNS xenograft failures (Czech et al., 1997). Taken together, these findings indicate that the CNS is accessible to immune cells and that local immune responses within the CNS are regulated by several mechanisms. It seems likely that some of these mechanisms help to limit immune responses, with vital consequences for the functioning of the healthy CNS. According to this view and the data summarized further on, it seems that the healthy CNS is hostile to immune activity (Flugel et al., 2001; Moalem et al., 1999b) and is destroyed by it, whereas immune activity is an essential requirement for the protection and maintenance of the damaged CNS.
ACUTE DEGENERATIVE CONDITIONS: DESTRUCTION BY THE ENEMY WITHIN
Acute mechanical or biochemical injury to the mammalian CNS often results in an irreversible functional deficit (Kalb, 1995; Schwab and Bartholdi, 1996) for several reasons, including the poor ability of injured axons to regrow and a destructive series of injury-induced events that result in the spread of damage to neurons that escaped the direct injury (Faden, 1993; Hauben and Schwartz, 2003; Povlishock and Jenkins, 1995; Yoles and Schwartz, 1998). This spread of damage is known as secondary degeneration. Attempts to promote CNS recovery have focused on two goals: (1) stimulation of regrowth (Caroni and Schwab, 1988; Cheng et al., 1996; Chong et al., 1999; Davies et al., 1997; Li et al., 1997; Miwa et al., 1997; Neumann and Woolf, 1999; Rapalino et al., 1998; Reier et al., 1992; Wang et al., 1998) and (2) neuroprotection, or the arrest of self-perpetuating degeneration (Basso et al., 1996; Bavetta et al., 1999; Beattie et al., 1997; Behrmann et al., 1994; Bethea et al., 1998; Blight, 1989; Constantini and Young, 1994; Crowe et al., 1997; Gruner et al., 1996; Moalem et al., 1999a; Sanner et al., 1994; Schwartz et al., 1999a, b ; Yong et al., 1998).
The extent of recovery, in the absence of intervention, is a function of the amount of tissue that escaped the initial injury minus the delayed neuronal loss due to secondary degeneration. It follows that the final outcome would be improved if fibers that were initially undamaged or only marginally damaged could be rescued from secondary degeneration. Recognition of this fact has led to the emphasis on neuroprotection in the approach to CNS therapy. The spread of damage is mediated by numerous factors, some of which have been characterized. Interestingly, however, similar factors were found to operate in different insults and across species, suggesting that the progression of damage might reflect a loss of control over mechanisms that regulate self-components that, though normally essential, contribute to the damage spread under pathologic conditions. The increased presence of infiltrating or activated immune cells that accompanies degeneration often led scientists and clinicians to conclude that these cells contribute to the pathology. As discussed below, it seems that this conclusion is an oversimplification (Schwartz and Hauben, 2002).
MACROPHAGES AND MICROGLIA IN CENTRAL NERVOUS SYSTEM INJURY: ARE THEY HELPFUL OR HARMFUL?
Studies of recovery after CNS injury have traditionally been based on the assumption that the CNS is a unique tissue whose postinjury behavior is governed by rules different from those underlying the recovery of other types of injured tissue, including the peripheral nervous system (PNS). Studies during the early 1980s showed, however, that although transected CNS axons fail to regenerate in their own degenerative environment, they can grow into transplanted peripheral nerve bridges (David and Aguayo, 1981). This key discovery indicated that at least some CNS axons are intrinsically capable of regeneration and that the hostility to growth emanates from their environment. From this starting point some studies began to focus on comparisons, both within and among species, between regenerative and nonregenerative nervous systems. This line of research established the concept that CNS regeneration failure may be attributable, at least in part, to an inability of the cellular components surrounding the injured axons to create a balanced environment capable of permitting and supporting growth. Among the factors found to be hostile to regrowth in adult CNS nerves are myelin-associated proteins (Cai et al., 2002; McKerracher et al., 1994; Qiu et al., 2002; Schnell and Schwab, 1990; Schwab and Caroni, 1988; Schwab and Thoenen, 1985; Shibata et al., 1998) and extracellular matrix proteins such as chondroitin sulfate proteoglycans (Rudge and Silver, 1990; Zuo et al., 1998). Accordingly, for regeneration to occur, these inhibitors must be masked, neutralized, or eliminated.
These and other discoveries prompted a series of studies whose findings were translated into strategies for the treatment of transected CNS axons, with the goal of rendering the axons capable of regeneration. As an example, the transplantation of intercostal nerve segments (peripheral nerves) into completely transected spinal cords of adult rats was shown to lead to partial regeneration, with restoration of hind limb function (Cheng et al., 1996). Monoclonal antibodies directed specifically against the myelin-associated inhibitor IN-1 were introduced into partially transected rat spinal cords, with consequent promotion of regeneration (Schnell and Schwab, 1990). More recently, inhibitors of signal transduction associated with myelin-related growth inhibitors were found to neutralize the inhibition of CNS regrowth (McKerracher, 2002; Qiu et al., 2002). Similarly, regrowth was promoted by treatment with proteases that degrade chondroitin sulfate proteoglycans (Bradbury et al., 2002; Rudge and Silver, 1990). Other approaches aimed at promoting recovery of CNS axons have been based on modification of the environment by local transplantation of embryonic tissue (Grady et al., 1985), Schwann cells (Xu et al., 1997), or cells that provide a source of trophic factors to increase the survival rate of the cell bodies (Coumans et al., 2001; Tuszynski and Gage, 1995).
Although immune cells are known to play a key role in tissue repair, their activity in the CNS, as outlined previously, has mostly been viewed as detrimental. A comparative study of the local inflammatory response in injured PNS and CNS axons showed that, in the injured PNS, where regeneration takes place spontaneously, both degeneration and regeneration appear to be brought about and controlled with the prominent participation of macrophages. Macrophages are also a source of cytokines and growth factors that actively participate, both directly and indirectly, in regrowth. As an example, the regeneration of optic nerves in lower vertebrates, as well as of peripheral nerves in mammals, was found to correlate with upregulation of macrophage-derived apolipo-proteins, which participate in the recycling of lipids needed for membrane rebuilding (Harel et al., 1989; Heumann et al., 1987; Ignatius et al., 1987). Similarly, the synthesis of nerve growth factor seems to be regulated by stimulated macrophages. Interleukin-1 and tumor necrosis factor-α, both secreted by macrophages, probably induce nerve growth factor transcription in Schwann cells (Lindholm et al., 1987, 1988).
Literature reports suggest that microglia in the intact CNS are in a downregulated form and, therefore, that their activation is likely to be restricted relative to that of macrophages which invade or reside in other tissues (Hailer et al., 1997; Neumann et al., 1996).
In early experiments aimed at overcoming the apparently inherent inadequacy (in terms of numbers and the timing and intensity of activity) of the innate response in the injured CNS, our research group found that implantation of homologous macrophages in the transected optic nerves of adult rats resulted in regrowth of the transected nerves (Lazarov-Spiegler et al., 1996). The observed regrowth was accompanied by other features known to be associated with peripheral nerve regrowth, such as the efficient clearance of dead cells and debris from the degenerating part of the nerve and the appearance of activated macrophages/microglia along the part of the nerve distal to the injury. After implanting activated macrophages in the completely transected spinal cords of adult rats, we showed partial recovery of motor function (manifested by locomotor activity scored in an open field) and electrophysiologic activity (assessed by motor-evoked potential responses) (Rapalino et al., 1998). In that study of injured spinal cords, the rats remained paralyzed for 2 months before showing the first signs of recovery. Approximately 60% of the rats showed recovery manifested by extensive movement of all hind limb joints with plantar placement of the paws. A few were even able to support their weight. Most of the control animals that were untreated or were treated only with the vehicle remained completely paralyzed, though some of them showed slight movement of more than two joints, without plantar placement. These behavioral manifestations were also reflected in the electrophysiologic recovery of motor-evoked potential responses in the implanted rats. The extent of recovery, in terms of the numbers of recovered muscles and bilaterality, varied among the rats (Rapalino et al., 1998). The effect on recovery was found to be a function of the macrophage dose (Rapalino et al., 1998).
Other studies have also provided evidence that microglia and macrophages can, under certain conditions, promote regeneration (Franzen et al., 1998; Prewitt et al., 1997). Conversely, there is evidence suggesting that the damaged spinal cord benefits from macrophage depletion (Popovich et al., 1999). These apparently contradictory findings might be attributable to differences in the animal strains in which the experiments were conducted, the injury model, and the measured outcome (Hauben and Schwartz, 2003; Kipnis et al., 2001; Schwartz and Kipnis, 2002a).
Close examination of the processes of neuronal regrowth and neuronal protection suggests that within the same process the requirements for recovery might vary at different postinjury stages. As an example, the antiinflammatory cytokine interleukin-10, which has a strong positive effect on recovery when applied soon after spinal cord injury (Bethea et al., 1999) is not beneficial when first applied at later posttraumatic stages. Moreover, observations of effective neuroprotective treatment, for example by steroids (Bracken, 1991; Hess and Sellon, 1997) or hypothermia (Chatzipanteli et al., 2000; Ghirnikar et al., 2000), have been interpreted to mean that inflammation is bad for recovery. Along the same lines, antiinflammatory treatment was found to be neuroprotective only if applied within the first few hours after the insult (Bracken and Holford, 1993; Young et al., 1994). In interpreting these findings, it should be borne in mind that the mere presence of activated microglia/macrophages cannot be taken as an indication of their deleterious effect. The activity of macrophages/microglia can however be both cytotoxic and protective (Shaked et al., unpublished observations, 2003) and is profoundly affected by the nature of the tissue and the presence or absence of other immune cells.
ADAPTIVE IMMUNITY AS A REGULATOR OF MICROGLIAL ABILITY TO MAINTAIN DAMAGED TISSUE
Our studies of macrophage participation in the recovery of injured CNS nerves led us to suggest that the fate of damaged nerves, like the fate of any damaged tissue, is determined by the immune system. Because the defensive role of the immune system in the rest of the body comprises both an innate (macrophage-mediated) and an adaptive (T cell- or B cell–mediated) response, we were interested in finding out whether the adaptive immune response also plays a role in the response to and the recovery from injury to the CNS and, if so, whether it operates by controlling the local innate response. Until we published our findings in this connection (Moalem et al., 1999a), the prevailing notion was that the adaptive immune response, represented by lymphocyte entry into the injured CNS, is likely to have only a negative effect on recovery. Given this view, it was surprising to find that CNS injury in rats is followed by an accumulation of T cells at the injury site, and that the T cells found to accumulate there in large numbers after passive transfer of any T cells have a protective effect on the remaining neurons (Hauben et al., 2000a; Moalem et al., 1999a, b ) (Fig. 1). In rats with spinal cord contusion, passive transfer of T cells specific to myelin basic protein (MBP; TMBP cells) led to a better recovery than that observed in control-injured rats without such T cell treatment. This finding was confirmed by locomotor activity of the rats in an open field and substantiated by morphologic and imaging criteria (Barouch and Schwartz, 2002; Butovsky et al., 2001; Hauben et al., 2000a, b ; Nevo et al., 2001). Besides the functional recovery, another dramatic effect was the dimension of cavity formation often seen in spinal cord injuries (Fig. 2).
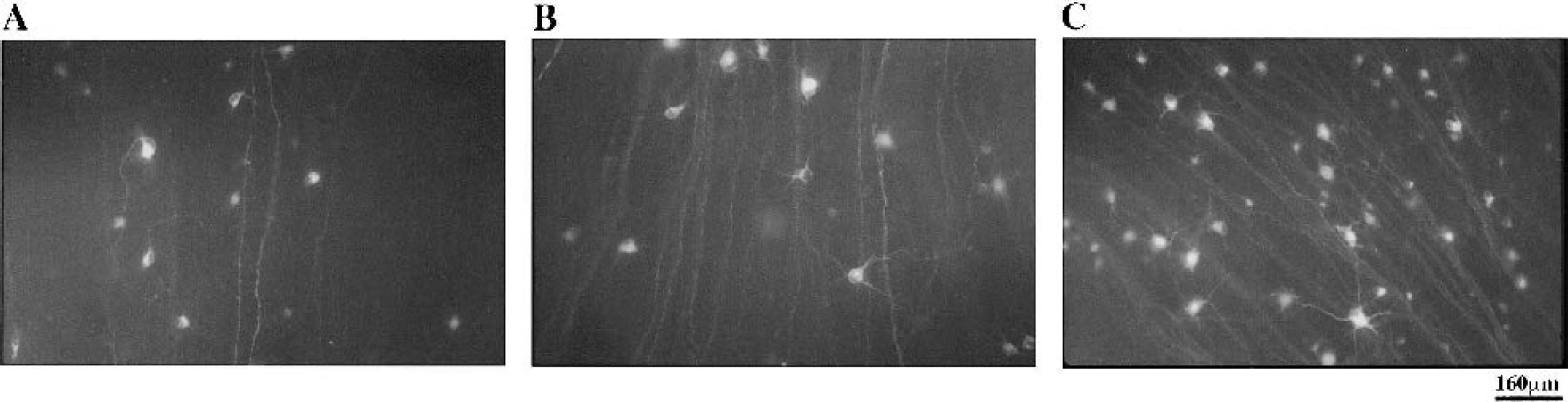
Photomicrographs of retrogradely labeled retinas of injured optic nerves of rats. Immediately after unilateral crush injury of their optic nerves, rats were injected with phosphate-buffered saline (controls)
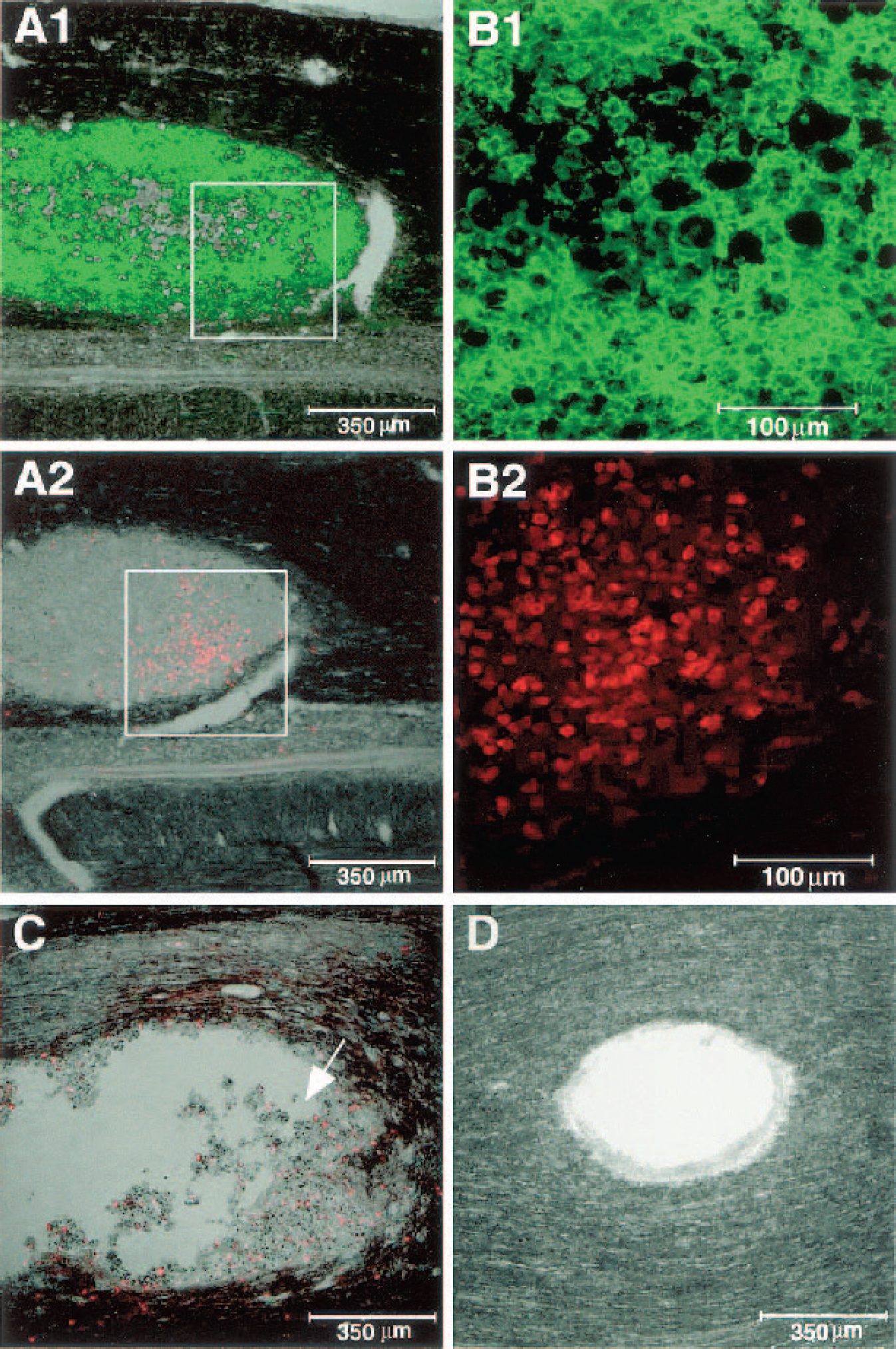
Colocalization of B7-2–expressing cells and T cells 28 d after injury in TMBP-treated and phosphate-buffered saline (PBS)-treated rats. Micrographs are from TMBP-treated rats
We further showed that the improvement in recovery observed in the spinally contused rats after passive T cell transfer can also be achieved by active immunization (vaccination) with myelin proteins, myelin-associated peptides or with altered myelin-derived peptides (Fig. 3) emulsified in the appropriate adjuvant (Hauben et al., 2000a, 2001).
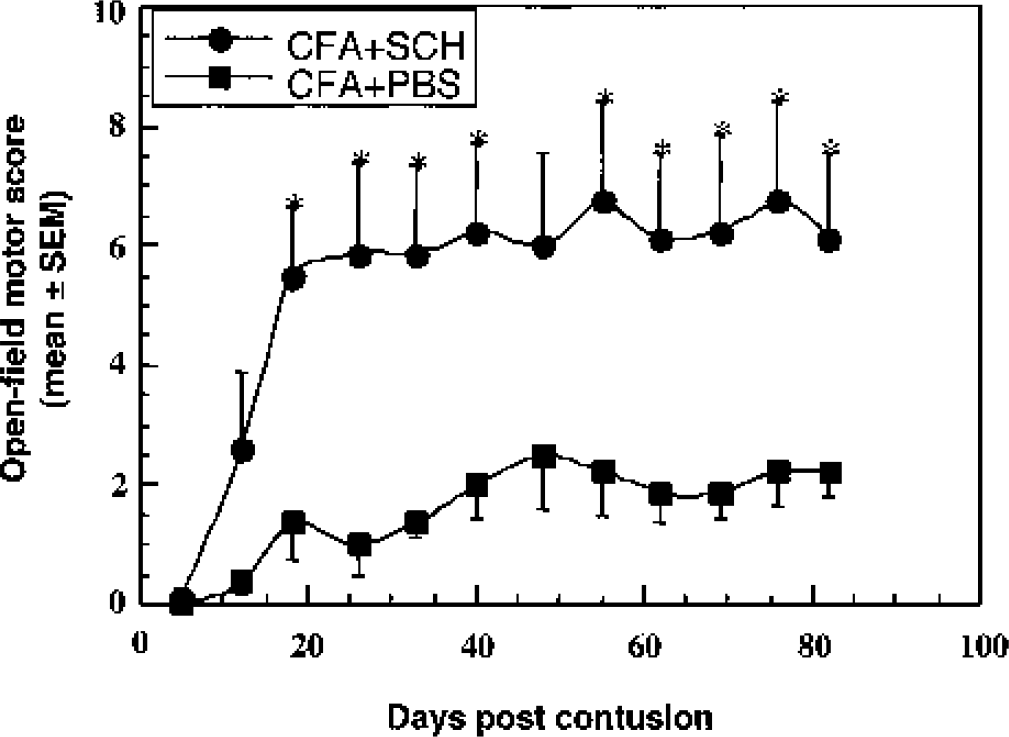
Functional outcome of spinal cord contusion in rats of a strain resistant to experimental autoimmune encephalomyelitis can be improved by active immunization. Five male Sprague-Dawley rats were immunized with spinal cord homogenate (SCH) emulsified in complete Freund's adjuvant (CFA; containing 0.5 mg/mL bacteria), and five were injected with phosphate-buffered saline (PBS) in the same adjuvant. Twelve days later the rats were subjected to spinal cord contusion, and their locomotor behavior in an open field was scored at the indicated times. Significantly better recovery was observed in the immunized rats than in the PBS-treated controls (P < 0.05, two-way ANOVA with replications; * P < 0.05, two-tailed Student's t test). (Reprinted with permission from J Clin Invest, vol. 108, Hauben E, Agranov E, Gothilf A, et al., Posttraumatic therapeutic vaccination with modified myelin self-antigen prevents complete paralysis while avoiding autoimmune disease, pp. 591–599, 2001.)
T CELL–MEDIATED PROTECTION: A PHYSIOLOGIC RESPONSE TO SPECIFIC ANTIGENS RESIDING IN THE SITE OF STRESS
The discovery that passive or active immunization with myelin-derived peptides can protect neurons from the consequences of axonal injury prompted us to examine whether the T cell–mediated protection can overcome the deleterious effect of factors known to play a prominent part in degenerative processes, such as glutamate or oxidative stress. The number of surviving neurons after exposure of rats to glutamate toxicity or oxidative stress was indeed lower if the rats were deprived of T cells (Kipnis et al., 2000; Schori et al., 2001a, b ). In seeking a suitable nonpathogenic antigen to use as a therapeutic vaccine for boosting this physiologic T cell–mediated protective response to the CNS insults, it was necessary first to find out the nature of the physiologic response. Studies from our laboratory showed that the physiologic T cell–based protection is directed against specific antigens residing in the site of the lesion (Kipnis et al., 2002a; Mizrahi et al., 2002; Yoles et al., 2001). The antigenic specificity is determined not by the type of the lesion but by its location (Mizrahi et al., 2002; Schori et al., 2001a). We interpreted this finding to mean that, to exert their protective effect, the T cells need to be activated locally. This can occur if the relevant antigens are presented to the T cells at the lesion site. Accordingly, after damage to myelinated axons the target antigens are proteins and peptides of myelin, whereas in the case of damage caused directly to the retina (for example by glutamate toxicity or an increase in intraocular pressure) the specific antigens are retinal proteins. The spontaneous response to an injury was indeed found to be evoked against the dominant antigens at the site of the lesion, as shown by the fact that if rats are “tolerized” by immunization with these immunodominant proteins at birth, their ability as adults to resist the consequences of CNS damage is wiped out (Kipnis et al., 2002a).
More recent studies led us to suggest that both the neuroprotective autoimmune T cells and the autoimmune T cells that cause an autoimmune disease are Th1 cells (Kipnis et al., 2002b). Moreover, while the phenotype and the antigenic specificity of the T cells are identical in both cases, protection can be achieved by both cryptic and pathogenic epitopes within the same antigen (Fisher et al., 2001; Moalem et al., 1999a), whereas disease is caused by the pathogenic epitopes only. Accordingly, we suggested that in degenerative conditions the default mechanism is protective autoimmunity, and that an autoimmune disease results only when the autoimmunity is malfunctioning (Schwartz and Kipnis, 2002b). The fact that autoimmunity was shown to be the body's repair mechanism and the finding that both disease and protection are mediated by the same cells raised some questions about the balance between the need for autoimmunity and the risk of autoimmune disease. Our most recent data suggest that the naturally occurring CD4+CD25+ regulatory T cells, which are responsible for avoiding autoimmune disease via suppression of peripheral autoimmunity, can preserve the balance by allowing the autoimmune T cells to exist in a state of alert in the healthy person, ready to be activated when needed for protection, while not causing autoimmune disease (Kipnis et al., 2002a; Kipnis et al., 2000; Schwartz and Kipnis, 2002a). Indeed, depletion of these regulatory T cells improves neuronal survival after CNS insult (Fig. 4).
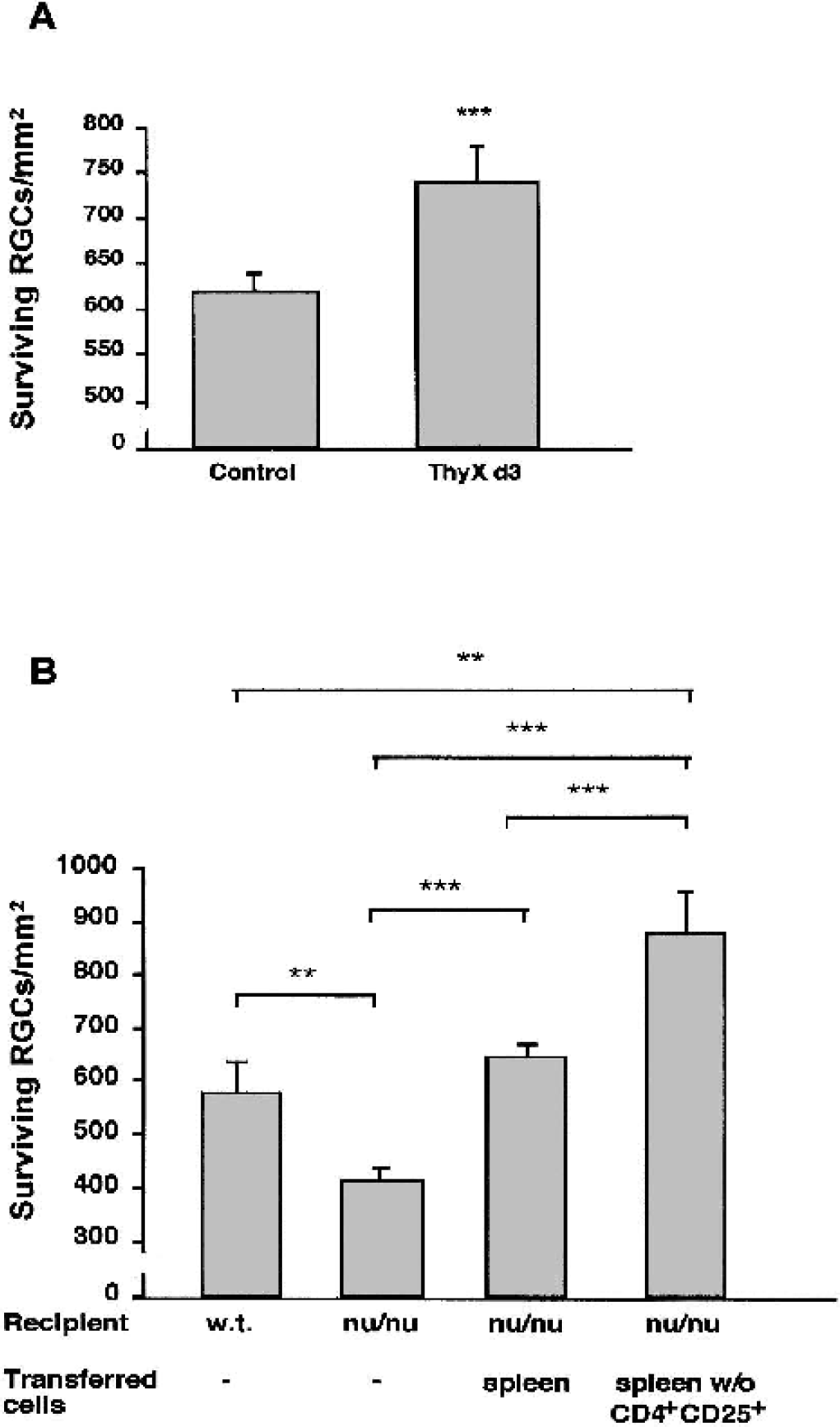
Depletion of naturally occurring regulatory CD4+CD25+ T cells in mice of an experimental autoimmune encephalomyelitis–susceptible strain improves neuronal survival after optic nerve crush injury.
POSSIBLE CONNECTION BETWEEN T CELL–BASED PROTECTION AND THE INNATE IMMUNE RESPONSE AT THE LESION SITE
Studies of recovery from CNS injury suggest that the target of the activated autoimmune T cells is the resident microglial population. Their activation by T cells allows the microglia to express dual activity: as immune cells with the function of antigen presentation (Butovsky et al., 2001) and as neural cells that supplement the buffering of destructive self-compounds. Passive transfer of T cells specific to myelin antigens, or active immunization with those antigens, results in a local response characterized by upregulation of B7.2 and MHC class II proteins (Butovsky et al., 2001). Preliminary data in our laboratory suggest that after microglia are exposed to activated T cells their capacity for glutamate uptake is increased. After such exposure the microglia apparently acquire an antigen-presenting phenotype, which facilitates their further communication with and activation of T cells. These findings suggest that microglia in the healthy CNS are on stand-by for activity when needed. In the case of CNS injury, they exercise their dual function in the stressed tissue (Shaked et al., unpublished observations, 2003). Other studies have shown that the infiltrating T cells, once activated, can serve as a source of neurotrophic factors (Moalem et al., 2000). These neurotrophic factors, or neurotrophic factor induced to be expressed by resident cells within the damaged nerve (Barouch and Schwartz, 2002) may contribute to the overall beneficial effect of the T cells in the healing process.
SYNTHESIZING THE FINDINGS
The T cell–mediated response directed to self-antigens residing in the site of CNS damage is spontaneously evoked. T cells isolated from the lymphocytes of spinally injured rats of Lewis, a strain susceptible to experimental autoimmune encephalomyelitis (EAE), when injected into naive syngeneic rats, are capable of causing neurologic deficits and histopathologic changes similar to those seen in EAE (Popovich et al., 1996, 2001). Our findings have shown that T cells isolated from spinally injured rats of a strain known to be resistant to autoimmune CNS diseases, when transferred into newly injured rats, are capable of conferring neuroprotection. We therefore suggest that an insult to the CNS, caused either by exogenous pathogens or by pathogen-free trauma, signals the immune system for assistance in protecting the tissue against the local threat. Experiments using different T cell lines in vitro suggest that the neuroprotection induced by weakly encephalitogenic and by strongly encephalitogenic autoreactive T cells is similar (Moalem et al., 1999a), supporting the notion that autoimmune T cells may be stimulated to facilitate protection of the damaged tissue against further damage, without inducing an autoimmune disease. The fact that the protection evoked by the spontaneous T cell response to a CNS insult is too weak to cause significant recovery might be attributable, at least in part, to the activity of the naturally occurring CD4+CD25+ regulatory T cells, whose presence might reflect an evolutionary compromise between two conflicting needs, namely for the presence of patrolling autoimmune T cells on alert for action in healthy persons and avoidance of the risk of autoimmune disease posed by these autoimmune T cells (Kipnis et al., 2002a; Schwartz and Kipnis, 2002a). This view of beneficial autoimmunity in the context of CNS trauma, supported by the work of other groups (Kerchensteiner et al., 1999; Olsson et al., 2003), might explain the presence of autoimmune T cells frequently observed in healthy persons. Our theory that inflammation, as the body's healing process, is mediated by microglia with the assistance of activated autoimmune T cells appears to contradict the conclusions of many of the studies on neurodegenerative diseases (Hauben and Schwartz, 2003; Schwartz and Kipnis, 2002a). The difference between our interpretation and those of others might be attributable to the fact that in most of the other studies the possibility that inflammation is a purposeful response in the CNS has been ignored. There are several possible reasons for this: (1) the fact that inflammation in general, and postinjury inflammation in particular, is not a single event but a series of events that together produce a multifactorial and multiparameter dialog between the tissue and the immune systems; (2) differences in the insult model selected for study (transection, contusion, injection of yeast, etc.); (3) strain differences (susceptible or resistant to autoimmune disease); (4) differences in the duration of the assessment period (short or steady state) and in the parameter assessed (morphologic or behavioral); (5) the fact that the beneficial effect of inflammation does not come free of charge (in terms of additional neuronal loss); (6) the need for an optimal balance in terms of the time of onset, duration, phenotype, and intensity of inflammation.
It is clear from our results that if the immune cells are not active enough, their numbers are insufficient, or their phenotype is inappropriate, the damage continues to progress in the presence of the activated innate immune cells, leading observers to assume that these were the cells causing the ongoing spread of damage (Eikelenboom et al., 2002; Lue and Walker, 2002; Magnus et al., 2002; McGeer and McGeer, 2002; Polazzi and Contesabile, 2002; Srebro and Dziobek, 2001; Taylor et al., 2002). Our data suggest, however, that the presence of activated immune cells at the lesion site does not necessarily mean that these cells are contributing to the ongoing damage. It seems more likely that they were recruited to help but are unequal to the task. We therefore envisage the following scenario: microglia, which can be either destructive or protective, are recruited to the lesion site. If they are properly activated but are not sufficiently numerous to be effective, the degeneration continues. This is what happens in the case of neurodegenerative disorders. If, on the other hand, the activation is not appropriate or if the immune system is malfunctioning, the autoimmune T cells might contribute to the ongoing degeneration. In the former case, boosting of the autoimmune response without risk of autoimmune disease will be beneficial. In our experiments with rats and mice, we have successfully adopted this approach as a therapeutic measure, using a weak self-antigen to make sure of activating only T cells with low affinity and avoiding pathogenic T cell clones.
THERAPEUTIC VACCINATION: BOOSTING THE BODY'S MECHANISM OF REPAIR
Our finding that the immune system participates in protection against destructive self-compounds by harnessing an anti-self immune response has been a milestone in the understanding of such self-compounds and of the nature and function of autoimmunity. Thus, until very recently, enemies of endogenous origin were not considered to be in need of attention by the adaptive immune system but were assumed to be taken care of by specific local buffering mechanisms (such as active transport of glutamate, or scavenging of free radicals). Autoimmunity was viewed as an attack by the body against itself. Our work put forward the novel ideas that “anti-self” is not less an immune defense mechanism than “anti-nonself,” and that, just as the immune system is called on to deal with invading microbes and other exogenous enemies, immune assistance is also needed for protection against the enemy within. Putting the two ideas together, we proposed that anti-self is the immune defense mechanism against harmful self-compounds. The self-enemies and the antigenic specificity are not identical because the antigenic specificity serves the role of homing, and thus any self-antigen expressed within the site of damage can serve the purpose of recruitment of an immune response to the site of damage in which self-enemies are invoked. Moreover, just as a defensive response against invading microbes can be boosted by vaccination with the appropriate foreign antigen, so it is possible to boost the anti-self defensive response by vaccination with a suitable self-antigen. Again, just as antimicrobial vaccines make use of attenuated foreign antigens, vaccination with weak self-antigens will be safer than the self itself. Our studies have indeed shown that vaccination with weak self or self-like compounds reduces the spread of degeneration in acute or chronic CNS degenerative conditions. Since glutamate is known to be a common player in many neurodegenerative conditions, finding a way to boost an immune response that is protective against glutamate may serve the purpose of protection against neurodegenerative diseases. We showed, for example, that glatiramer acetate, also known as copolymer 1, can protect rats and mice against several adverse conditions in the CNS (Bakalash et al., unpublished data, 2003) (Angelov et al., 2003; Kipnis et al., 2000; Schori et al., 2001a). Further studies are needed to optimize the protocol for treatment in various chronic conditions and to differentiate it from that used in patients with a chronic autoimmune disease (Kipnis and Schwartz, 2002).