
Abstract
The 70-kDa heat shock protein (Hsp70) is involved in protecting the brain from a variety of insults including stroke. Although the mechanism has been largely considered to be because of its chaperone functions, recent work indicates that Hsp70 also modulates inflammatory responses. To explore how and whether Hsp70 regulate immune responses in brain ischemia, mice overexpressing Hsp70 (Hsp Tg) were subjected to 2 h middle cerebral artery occlusion, followed by 24 h reperfusion. Parallel experiments were performed using a brain inflammation model. Hsp Tg microglia cocultured with astrocytes were used to evaluate the direct effects of Hsp70 on cytotoxicity of mcrigolia. Compared with wild-type (Wt) littermates, Hsp Tg mice showed decreased infarct size and improved neurological deficits. The number of activated microglia/macrophages were also reduced in ischemic brains of Hsp Tg mice. Similar observations were made in a model of brain inflammation that does not result in brain cell death. Overexpression of Hsp70 in microglia completely prevented microglia-induced cytotoxicity to astrocytes. Activation of the inflammatory transcription factor, nuclear factor-κB (NF-κB) was inhibited significantly in Hsp Tg mice and microglia. This was associated with decreased phosphorylation of NF-κB inhibitor protein, IκBα, and decreased expression of several NFκB-regulated genes. Co-immunoprecipitation studies revealed an interaction of Hsp70 with NF-κB and IκBα, but not with IkB kinase, IKKγ, suggesting that Hsp70 binds to the NF-κB:IκB complex preventing IκB phosphorylation by IKK. The findings of the present work establish an anti-inflammatory role for Hsp70 in the context of brain ischemia as a novel mechanism of protection.
Introduction
The 72-kDa heat shock protein (Hsp70) is a highly stress-inducible member of a chaperone family of proteins (Mayer and Bukau, 2005). Work by our group and others showed that overexpressing Hsp70 is protective against focal and global cerebral ischemia and kainic acid-induced neurotoxicity (Kelly et al, 2002; Plumier et al, 1997; Rajdev et al, 2000; Tsuchiya et al, 2003; Yenari et al, 1998). Conversely, Hsp70.1 deficiency leads to worsened outcome after experimental stroke (Lee et al, 2001b). The mechanism of protection after cerebral ischemic events is not well known, but has largely been attributed to its chaperone functions whereby HSP70 improves cell survival by preventing protein aggregation (Giffard et al, 2004). A few studies suggest that HSP70 may protect via antiapoptotic mechanisms (Matsumori et al, 2005, 2006; Yenari et al, 2005). Recent work has shown that heat shock proteins (Hsps) are capable of modulating immune responses either by potentiating or inhibiting them, but this has never been addressed in brain ischemia or injury. As it is fairly well recognized that inflammation participates in cerebral ischemic injury by contributing to brain tissue damage acutely, we explored whether and how Hsp70 modulates inflammatory responses in a model of transient middle cerebral artery occlusion (tMCAO) in transgenic mice overexpressing Hsp70. In addition, as infarct size might predict the extent of the post-ischemic inflammation, parallel experiments were performed in a brain inflammation model, in which we showed previously does not cause detectable brain cell death (Deng et al, 2003), as well as in an in vitro model of ischemia-like injury. We found that overexpression of Hsp70 not only protects against brain ischemia but also appears to do so via an anti-inflammatory mechanism.
Materials and methods
Animals
Experiments were performed according to the animal protocols approved by institutional panels on laboratory animal care. Male Hsp70 transgenic mice (Hsp Tg) and wild-type (Wt) littermates originally created by Dillmann et al (University of California, San Diego; Marber et al, 1995) were used. Rat-inducible Hsp70 was placed under the control of β-actin promoter and cytomegalovirus enhancer. Transgenic mice were bred with mice of the same strain (B6 × SJL) to generate heterozygote animals. The genotypes of the resultant litters were confirmed with polymerase chain reaction analysis of DNA extracted from the tails. Overexpression of Hsp70 was showed by Western blot analysis. After backcrossing for five generations, male adult Hsp Tg and Wt mice weighting 25 to 28 g were used for this study.
Transient Middle Cerebral Artery Occlusion Model
Ischemia was induced using the tMCAO model as described previously (Lee et al, 2001a). Briefly, mice were anesthetized by face mask using 3% to 4% isofluorane followed by 1.5% to 2% for maintenance. Physiological parameters including body temperature, blood pressure, and arterial blood gases were monitored and maintained in the normal range. A 6-0 nylon monofilament suture was inserted into the common carotid artery to occlude the ostium of middle cerebral artery. After 2 h, the suture was retracted to allow reperfusion. Focal neurological deficits were evaluated at 2 and 24 h after MCAO using a neurological score based on that developed by Bederson et al, (1986) and modified for use in mice (Rajdev et al, 2000): grade 0 = no observable neurological deficits, grade 1 = fails to extend right forepaw, grade 2 = circles to the right, grade 3 = falls to the right, and grade 4 = cannot walk spontaneously. Animals with no observable deficits at the time of recovery from anesthesia were removed from the experiment. Twenty-four hours later, the animals were euthanized in a CO2 chamber and then perfused transcardially with heparinized saline. Brains were harvested for different assays.
Brain Inflammation Model
Anesthetized mice were injected intravenously with 2.5 mg/kg of lipopolysaccharide (LPS, Escherichia coli serotype 055:B5; Sigma, St Louis, MO, USA) dissolved in sterile 0.9% saline. We showed previously that this model causes transient aseptic meningitis with inflammatory cell activation without any detectable brain cell death (Deng et al, 2003). Mice were euthanized 12 h after the injection.
Infarct Size
After animals were perfused, brains were sunk in 20% sucrose, frozen, and cut into 20 μm sections on a cryostat. Brain sections from four consecutive coronal levels of the brain were stained with cresyl violet. Areas of infarction were measured using an image analysis system, as previously described (Lee et al, 2001a).
Histochemistry and Fluorescent Microscopy
Microglia were identified using lectin histochemistry. Brain sections were stained with peroxidase labeled Griffonia simplicifolia isolectin-B4 (IB4) (10 μg/mL, GSAI-B4; Sigma, L5391) in phosphate-buffered saline (Han et al, 2002; Wang et al, 2002), followed by diaminobenzidine (Vector Laboratories, Burlingame, CA, USA), then counterstained with hematoxylin and eosin. For immunohistochemistry and fluorescent microscopy, brain sections were blocked, incubated in primary antibodies overnight at 4°C, followed by a biotinylated anti-rat IgG (1:2,000; Vector Laboratories) or Cy3-conjugated goat anti-rabbit IgG(H + L) (1:200, 56,399; Jackson Immuno Research, West Grove, PA, USA) at room temperature for 1 h, respectively (Han et al, 2003). For immunohistochemistry, proteins were detected using the Elite Vectastain ABC kit (Vector Laboratories) and diaminobenzidine, then counterstained with hematoxylin and eosin. For fluorescent immunostaining, sections were counterstained with the nuclear marker 4′,6′-diamidino-2-phenylindole (Vector Laboratories) and images were viewed with an epifluorescence microscope (Nikon Diaphot, Nikon Corporation, Melville, NY, USA). The following primary antibodies/concentrations were used: MHC Class II antibody (10 μg/mL, ab3084; Abcam, Cambridge, MA, USA); NF-κB p65 subunit (1:50, NF-κB p65, sc-109; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA). Brain regions selected for cell counting was based on procedure previously published by our group (Han et al, 2003; Wang et al, 2002). For tMCAO, positive cells were counted in a masked fashion in five random non-overlapping fields within the cortex adjacent to the outer boundary of the infarct, as delineated by the cresyl violet-stained sections. For the LPS-treated animals, positive cells were sampled from six adjacent fields in both lateral cortices for each animal. Counts were performed by an investigator masked to the conditions of the experiment.
Cell Culture
Primary astrocyte or microglia cultures were prepared separately from brains of 1 to 3-days-old transgenic pups and Wt littermates, based on the protocol described previously by our group (Yenari and Giffard, 2001; Yenari et al, 2006). Astrocytes were isolated from postnatal (P1-3) mice and used for experiments 3 weeks later. Microglia were isolated from mixed glial cultures. Mixed glial cultures were grown in a flask in medium containing 10% serum. After 2 to 3 weeks, microglial cells could be seen floating on top of the confluent cell layer. Microglial cells were collected by shaking the culture flask at 80 r.p.m. for 20 mins. Microglial cells were seeded on top of confluent astrocytes cultures at a density of 2 × 105 cells/mL, and allowed to rest for 48 h before using in experiments.
Oxygen and Glucose Deprivation
Cultures of either astrocytes alone or astrocytes with microglial cells were deprived of glucose and oxygen by changing the culture medium to balanced salt solution containing no glucose (BSSO) and kept in an oxygen-free chamber at 37°C for 6 h. Cultures were then transferred to a 37°C incubator with 5% CO2 and reperfused with glucose at a concentration of 5.5 mmol/L (BSS5.5) at normoxia for 24 h. All experiments were performed in triplicate.
Cell Death Assay
Cell death was quantified by staining with trypan blue and by assay of released lactate dehydrogenase released into the media (Lee et al, 2001a), expressed as a percent of the total lactate dehydrogenase released after freeze-thaw (= 100%). As only astrocytes (and not microglia) die after 6 h OGD (Yenari and Giffard, 2001), trypan blue-positive cells and lactate dehydrogenase signal was due to astrocyte death.
Western Blot Analysis
Whole cell lysates of brain tissue were prepared as described previously (Han et al, 2003). Cytoplasmic and nuclear protein subfractions were prepared as described previously (Dignam et al, 1983). Primary microglia were harvested using a trypsinization method to increase yield (Saura et al, 2003). Brain and cell lysates were prepared and suspended in lysis buffer containing sodium dodecyl sulfate and protease inhibitors. Denatured protein extracts (50 μg) from ipilateral or contralateral hemispheres or primary microglia were separated on a 10% Tris—HCl Ready Gel (Catalog No. L040605A2; Bio-Rad Laboratories, Hercules, CA, USA) and transferred to polyvinylidinene fluoride membrane (IPVH00010; Millipore, Bedford, MA, USA). The membranes were incubated with the antibodies below and the immunoreactive protein bands were visualized with enhanced chemiluminescence detection (Amersham Pharmacia Biotech, Piscataway, NJ, USA). Primary murine antibodies used were anti-Hsp70 (1:1,500, SPA-810, StressGen Biotechnologies), anti-IκBα (inhibitor of NF-κBα) (1:200, sc-1643; Santa Cruz Biotechnology Inc.), IgG2b p-IκBα, which recognizes only the phosphorylated form of IκBα (1:200, sc-8404; Santa Cruz Biotechnology Inc.), NF-κB p65 (sc-8008; 1:200, Santa Cruz Biotechnology Inc.), β-actin (A-5441, 1:5,000; Sigma), and anti-histone H1 Ab (1:1,000, sc-10806, Santa Cruz Biotechnology Inc.). Goat anti-mouse IgG HRP (1:1,000, sc-2055, Santa Cruz Biotechnology Inc.) was used as a secondary antibody. Densitometric measurements were made from films using Typhoon 9410 Variable Mode Imager (GE Health Care, Piscataway, NJ, USA), then quantified using ImageQuant 5.2 (Molecular Dynamics, Piscataway, NJ, USA). The amount of protein, normalized to actin as a reference, was expressed as relative intensity.
Microwell Colorimetric Nuclear Factor-κB p65 Assay
TransAM NF-κB p65 Transcription Factor Assay Kits (Active Motif, Carlsbad, CA, USA) was used to estimate the DNA-binding capacity of NF-κB, and was performed following kit instructions with minor alterations for brain tissue (Han et al, 2003). This method has been shown to be more sensitive than electrophoretic mobility shift assays (Shen et al, 2002). The optical density (OD) of the samples was quantified on a spectrophotometer at 450 nm with a reference wavelength of 655 nm. HeLa whole cell extracts were used as positive control and the NF-κB Wt and mutated consensus oligonucleotide were used to monitor the specificity of the assay. Nuclear factor-κB activity was expressed as a percentage by using the formula: (sample OD value—blank OD value)/positive control OD value.
Co-immunoprecipitation
This was performed by following a protocol from Stressgen Biotechnologies (Catalog and Technical Reference Guide) with minor modifications. Whole brain tissue lysates were pre-cleared by adding 50 μL Protein A/G PLUS-Agarose (sc-2003; Santa Cruz Biotechnology Inc.), 2 mg of tissue lysate in 1 mL of complete RIPA buffer. Precleared lysates (200 μL) were then incubated with 2.5 μg of mouse monoclonal Anti-Hsp70 antibody (SPA-810; Stressgen Biotechnologies) or an IgG isotype control (2.5 μg normal mouse IgG, sc-2025; Santa Cruz Biotechnology Inc.) at 4°C overnight. The Protein A/G PLUS-Agarose was then collected and the supernatant was aspirated off by microcentrifuging the mixture for 2 mins at 4,300g. After washing all reactions five times, samples were boiled for 5 mins and then microcentrifuged briefly to pellet Protein A/G PLUS-Agarose. Twenty microliters of the supernatants, precleared lysates, and non-precleared lysates were used for Western blot analysis. Antibodies used were purchased from Santa Cruz Biotechnology Inc. (primary antibodies: IκB (1:200, sc-1643), IKKγ (1: 1,000, sc-8330), NF-κB p65 (F-6) (1:200, sc-8008); secondary antibodies: goat anti-mouse IgG HRP (1:1,000, sc-2055); goat anti-rabbit IgG HRP (1:1,000, SC-2054). Co-immunoprecipitation was also performed in the reverse order by immunoprecipitating for the NF-κB/NF-κB-associated antibody followed by a Western blot of Hsp70.
RNA Preparation and TaqMan Real-Time Polymerase Chain Reaction
Brains tissue from the ipsilateral ischemic and contralateral hemispheres and hemispheres of LPS-treated mice were cut into 0.5 cm thick slices, and immediately submerged in the RNAlater RNA Stabilization Reagent (QIAGEN, Cat No. 76,106, Valencia, CA, USA). Total RNA was isolated by RNeasy® Lipid Tissue Midi Kit (QIAGEN, USA, Cat No.75,842). Synthesis of cDNA from total RNA samples was performed by using TaqMan® Reverse Transcription Reagents (Applied Biosystems, N808-0234). For each brain sample, 2 μg of total RNA was used in 100 μL of reverse transcription reaction in a 0.2 mL microtube.
Mouse cDNA sequences for TNF-α, IL-1β, ICAM-1, and inducible nitric oxide synthase (iNOS) was obtained from GenBank. The corresponding primers and TaqMan probes were designed with Primer Express 1.0 Software program (PE Applied Biosystems, Foster City, CA, USA), and synthesized by PE Applied Biosystems. FAM™ was used as the 5′-reporter dye and TAMRA™, as 3′-quencher dye of TaqMan probe. The DNA sequences of the primers and probes used were as follows: (1) TNFα: Fwd: 5′-GCCACC ACGCTCTTCTGT-3′, Rev: 5′-GGAGGCCATTTGGGAACT-3′, Intern: 5′-6FAMTACTGAACTTCGGGGTGA TCGGTCT AMRA-3′; (2) ICAM-1: Fwd: 5′-GCGCTTCTTTTGCTCTGC-3′, Rev: 5′-CCAAGC AGTCGGTCTCGT-3′, Intern: 5′-6FAMAG GTGGCGGGAAAGTTCCTGTCTATAMRA-3′; (3) iNOS: Fwd: 5′-CCCTGCTTTGTGCGAGT-3′, Rev: 5′-CCCAATGAGAT GCAAGG-3′, Intern: 5′-6FAMTGGCACATTCTGTTCAAA GAGAGCCTAMRA-3′; (4) IL-1β: Fwd: 5′-AGGTCGCTCA GGGTCACA-3′, Rev: 5′-GTGGTTGCCCATCAGAGG-3′, Intern: 5′-6FAMTGGCACATTCTGTTCAAAGAGAGCC TAMRA-3′.
Real-time polymerase chain reaction was performed using 9700 HT Fast Real-Time PCR System (Applied Biosystems). A mouse GAPDH endogenous control was used as an active reference to normalize quantifitation of targets for differences in the amount of total RNA added to each reaction. TaqMan® Universal PCR Master Mix (Applied Biosystems) was used for real-time detection of cDNA. Thermal cycling condition comprised 2 mins at 50°C, 10 mins at 95°C; 40 polymerase chain reaction cycles, each consisting of 15 secs at 95°C, 1 min at 60°C. The data were acquired and analyzed with Sequence Detector 2.0 software (Applied Biosystems). The amount of target, normalized to GAPDH endogenous reference and relative to a calibrator, is given by the arithmetic formula 2T−ΔΔC; data output was expressed as a fold difference of expression levels.
Statistical Analysis
SigmaStat (SPSS, Chicago, IL, USA) was use to analyze data. For both the ischemia and inflammation models, differences between groups were determined using Student's t-test, one-way analysis of variance for continuous data, and Mann—Whitney for non-continuous data. Statistical significance was determined at the P <0.05 level. All data are expressed as mean ± s.e.m.
Results
Overexpression of Hsp70 in Transgenic Mice
To confirm the overexpression of Hsp70 gene in Hsp Tg mice, Western blots were performed which show more than 10-fold higher expression of Hsp70 in the brain (Figure 1A), and in isolated microglia (Figure 1B) in Hsp Tg mice compared with Wt mice.
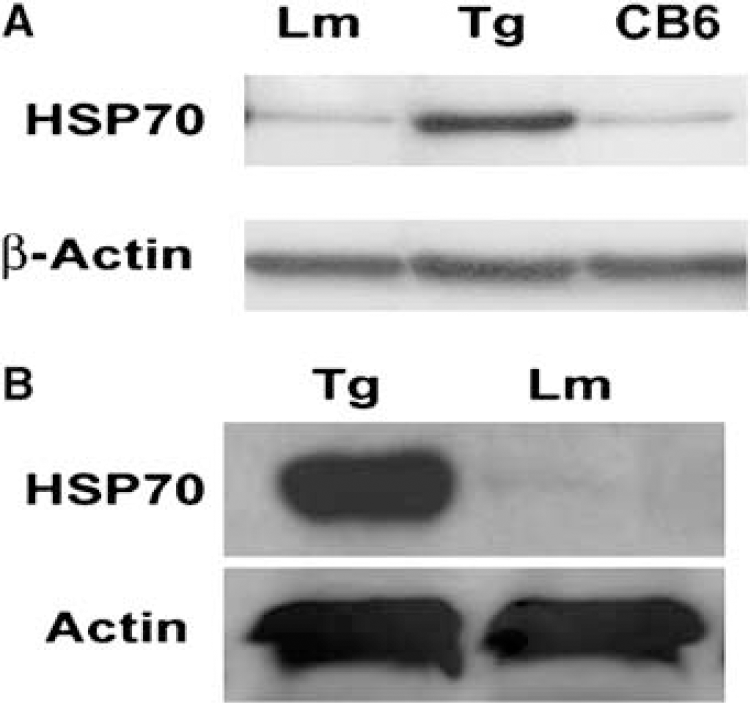
Overexpression of Hsp70 in transgenic mice. Western blot analysis shows ∼10-fold higher expression of Hsp70 protein in the brain of a Hsp70 transgenic mouse (Hsp Tg) compared to brains of either a wild-type littermate (LM) or background strain (CB6) (
Hsp70 Reduces Infarct Size and Improves the Neurological Deficits
We first established whether Hsp70 overexpression is neuroprotective in our hands. As shown in Figure 2A, a representative brain section from an Hsp70 Tg mouse has smaller infarct size than that from a Wt littermate. Total infarct size among Hsp70 Tg mice was significantly reduced by about 50% compared with Wt littermate mice (Figure 2B). Neurological scores were also improved among Hsp70 Tg mice compared with Wt mice at 24 h after MCAO (Figure 2C). These observations are in line with those made by others using this same mouse model (Rajdev et al, 2000; Tsuchiya et al, 2003).
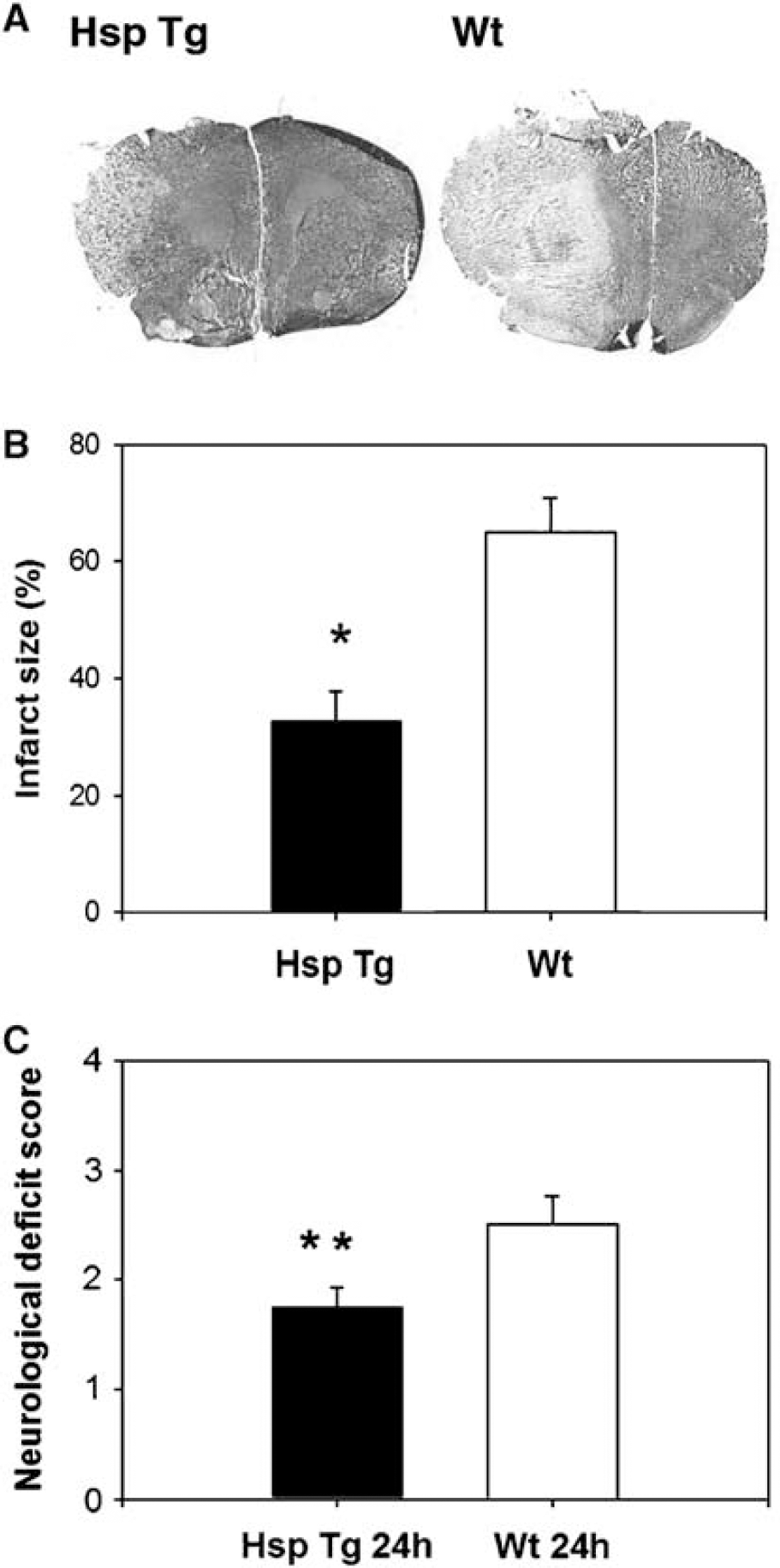
Reduced infarct size and improved neurological deficits among Hsp70 transgenic mice. (
Hsp70 Reduces Number of Active Microglia/Macrophages
Activated microglia and/or macrophages were visualized by morphological transformation, increased lectin reactivity, and MHC class II expression. We quantified the number of activated microglia (assessed by IB4 and MHC-class II staining) in the peri-infarct region within the ipsilateral ischemic cortex. Regions of interest were selected based on criteria published previously (Wang et al, 2002). In this study, we did not attempt to distinguish microglia from macrophages, taking into account the fact that they are virtually indistinguishable under pathological settings. Figure 3A shows more intensely stained IB4-positive cells in a brain section of a Wt littermate compared with an Hsp Tg brain 24 h after MCAO. Similarly, there were fewer MHC Class II-positive cells among Hsp Tg mice (Figure 3B). The number of activated microglia/macrophages as assessed by IB4 and MHC Class II immunostains were significantly decreased among the Hsp Tg mice compared with Wt (Figure 3C). A parallel study in the brain inflammation model also showed that the number of positive IB4 were decreased in Hsp Tg brains after LPS administration (Figure 3D).
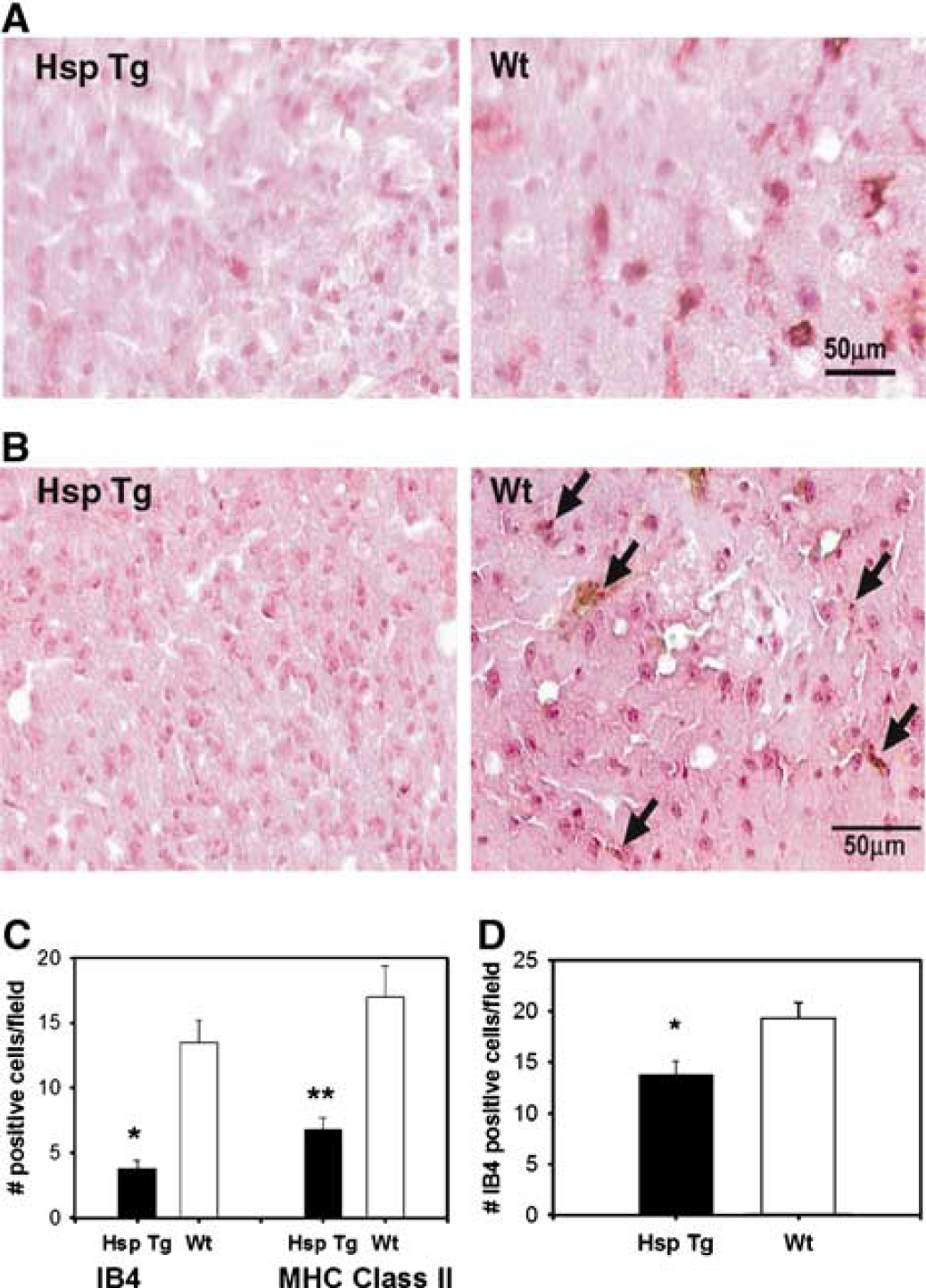
Hsp70 overexpression inhibits microglia/macrophage activation. (
Hsp70 Prevents Microglia-Induced Astrocyte Death In Vitro
To provide direct evidence of an anti-inflammatory effect of Hsp70, we then evaluated the effects of Hsp70 on microglia-induced astrocyte death in mixed microglia/astrocyte cultures after OGD. As shown in Figure 4, Wt microglia increases astrocyte death after OGD, whereas microglia-induced astrocyte death was completely inhibited when cultured with microglia from Hsp Tg mice.
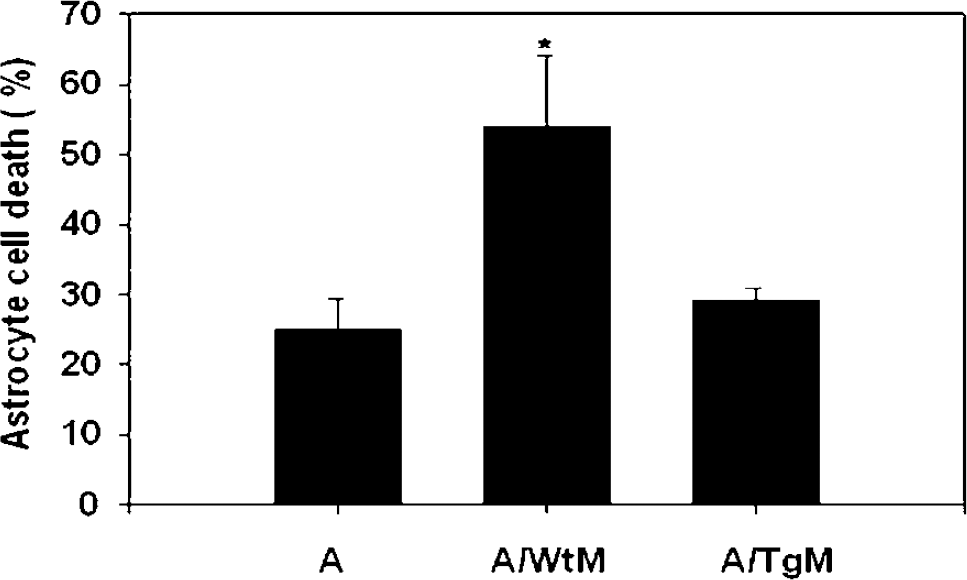
Hsp70 overexpression prevents microglia-induced astrocyte death. The addition of wild-type microglia (wt) to astrocyte cultures (astrocyte/WtM) increased cell death after OGD, compared with astrocytes cultured alone (A). Overexpression of Hsp70 in microglia prevented microglia-induced cell death after OGD. A, astrocytes alone; A/WtM: co-culture of wildtype astrocytes with Wt microglia; A/TgM, co-culture of wildtype astrocytes with Hsp Tg microglia. Mean±s.e. of three experiments is shown for each condition (*P < 0.001).
Hsp70 reduces nuclear Nuclear Factor-κB
A representative ischemic brain of a Wt mouse shows several cells with both cytosolic and nuclear staining of NF-κB p65 subunit, whereas that of a Hsp Tg mouse reveals predominately cytosolic NF-κB staining, with little to no staining in the nucleus (Figure 5A). The proportion of ischemic cells with nuclear NF-κB staining was significantly reduced among Hsp Tg mice (Figure 5B). These findings were further confirmed by performing Western blots of cytosolic and nuclear subfractions. Nuclear factor-κB was found in the nuclear subfractions of ischemic Wt brains. In contrast, NF-κB was unchanged in ischemic Hsp Tg brains with little to no nuclear expression (Figure 5C). Immunostains of NF-κB in cultured primary microglia after OGD revealed both cytosolic and nuclear staining in Wt cultures, but fewer cells with nuclear staining among Hsp Tg cultures (Figure 5D).
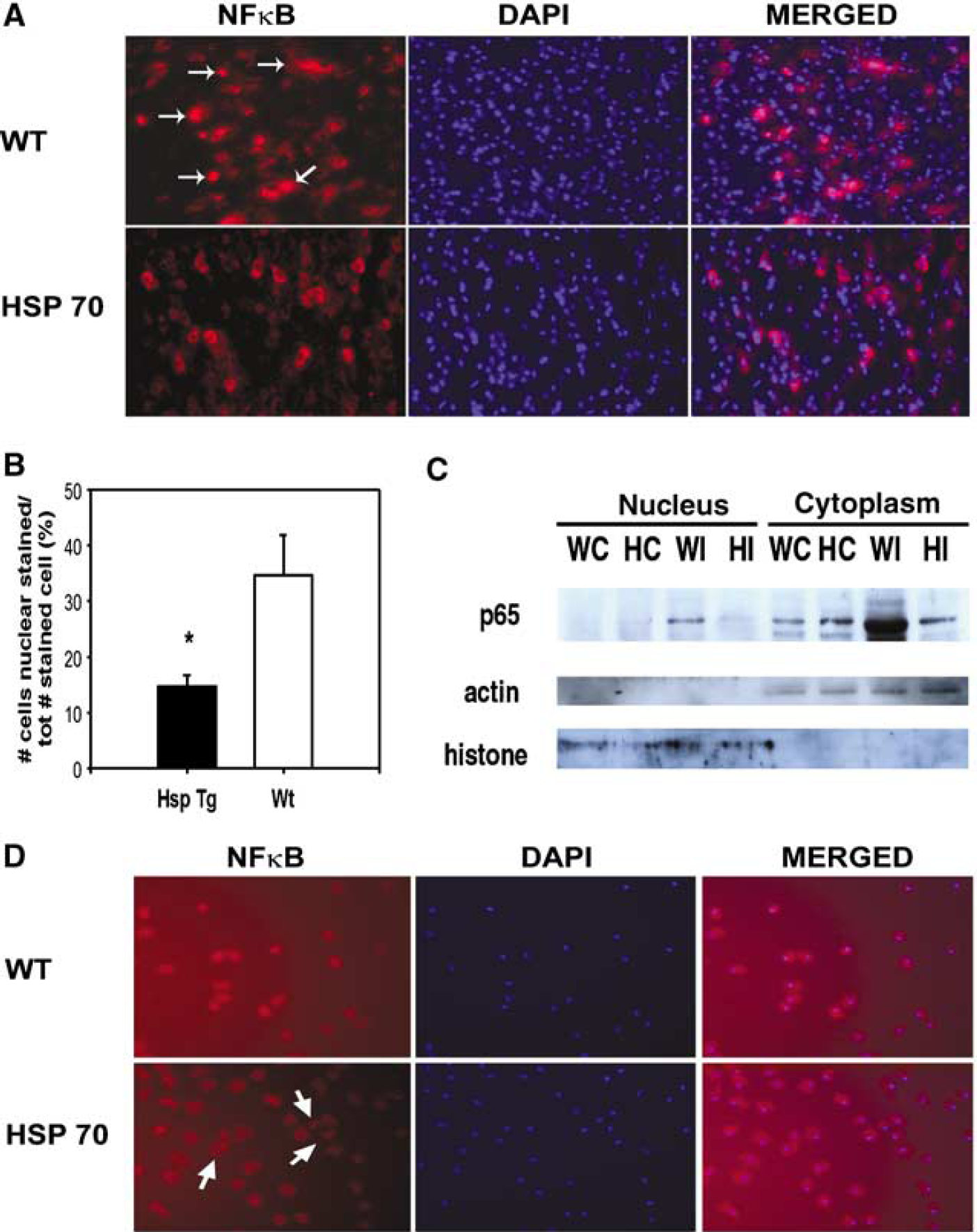
Hsp70 overexpression inhibits nuclear translocation of NFκB. (
Hsp70 Decreases Nuclear Factor-κB DNA Binding Capacity
To corroborate the inhibitory effects of Hsp 70 on NF-κB activation, we further examined NF-κB DNA binding capacity using a multiwell colorimetric technique that was designed to detect the ability of active NF-κB to bind to double-stranded oligonucleotide probes containing consensus binding sequences (5′-GGGACTTTCC-3′). This method was reported to be more sensitive and quantitative than classic electrophoretic mobility shift assay (Shen et al, 2002), and has been successfully applied to the brain tissue in our laboratory before (Han et al, 2003). Figure 6 shows that NF-κB activity among Hsp Tg mice is significantly decreased in both the stroke (Figure 6A) and brain inflammation (Figure 6B) models.
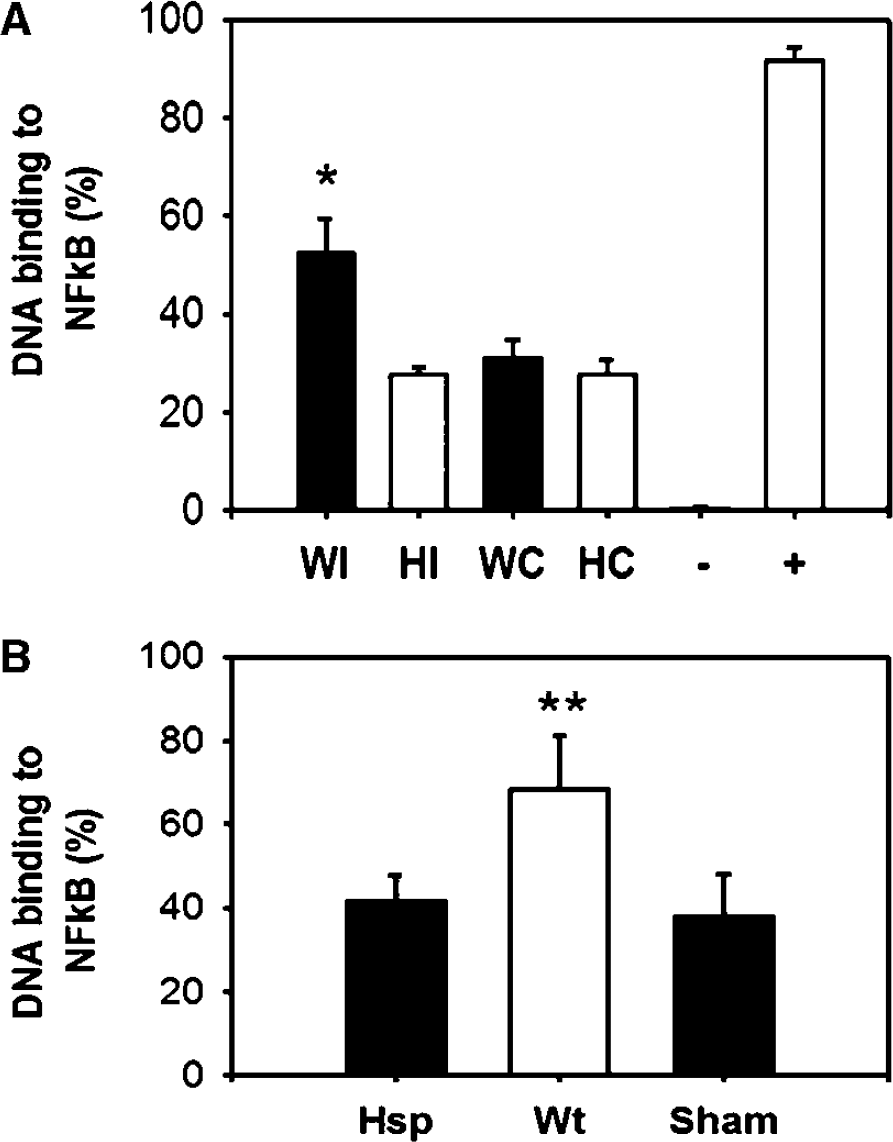
Decreased NFκB activity among Hsp70 transgenic mice. (
Hsp70 Inhibits IκB Phosphorylation
Phosphorylation of IκBα leads to subsequent IκBα degradation, thus liberating NF-κB to enter the nucleus. Western blots of IκBα, NF-κB p65, and phosphorylated IκBα (p-IκBα) after tMCAO revealed no significant differences in the amount of overall IκBα protein and NF-κB p65 between Hsp Tg and Wt mice (Figure 7). Uninjured Hsp Tg brains had no change in these same proteins compared with Wt (data not shown). However, phosphorylated IκBα levels were significantly decreased among Hsp Tg mice (Figure 7A). Similar patterns were observed in brain inflammation model (Figure 7B) suggesting that Hsp70 is somehow capable of preventing IκBα phosphorylation.
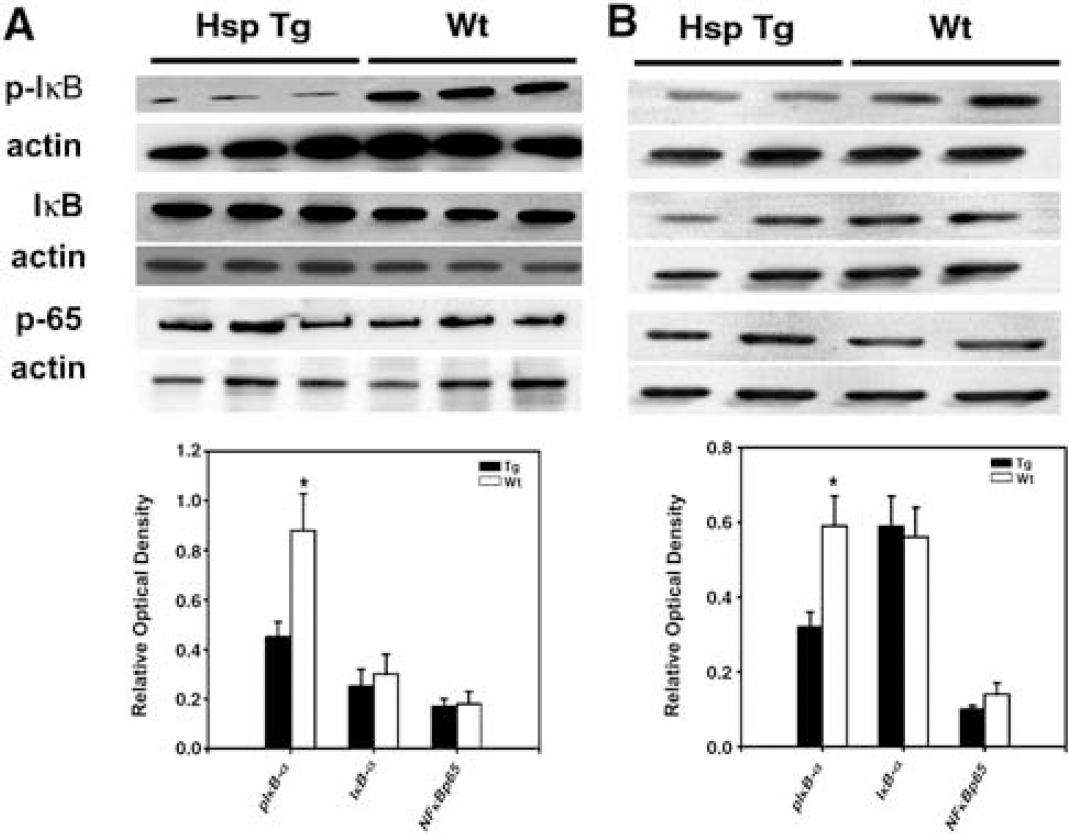
Less IκBα phosphorylation observed among Hsp70 transgenic mice. Western blots of IκBα and NFκB p65 show no difference in the amount of total IκB protein and NFκB p65 in the brains of Hsp Tg and Wt mice exposed to tMCAO (k) or LPS (
Hsp70 associates with NF-κB:IκB Complex
To explore further interactions between Hsp70 and the NF-κB complex, we performed co-immunoprecipitation experiments with IkB and IKKγ (IκB kinase). Hsp70 appeared to co-immunoprecipitate with NF-κB p65 subunit and IκBα, but not with IKKγ (Figure 8). Similar patterns were observed regardless of the order of the antibodies (data not shown). Similar results were observed in both the tMCAO and brain inflammation models.
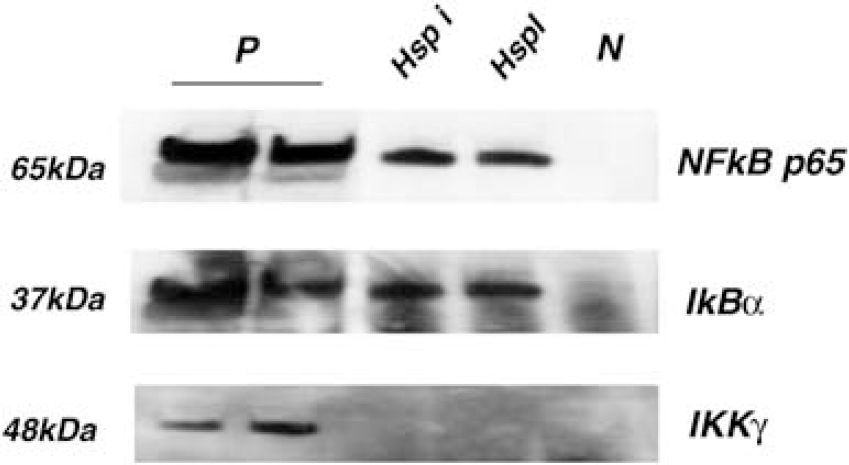
Hsp70 co-immunoprecipitates with the NFκB:IκB complex. Whole cell lysates were co-immunoprecipitated with Hsp70 antibody, followed by antibodies against NFκB's p65 subunit, IκBα or IKKγ. Hsp70 was found to associate with NFκB p65 and IκBα, but not with IKKγ following brain ischemia and endotoxin exposure (Hsp i, HspTg mice exposed to tMCAO; Hsp I, Hsp Tg mice exposed to LPS; P, protein lysates from Hsp i and Hsp I that were not immunoprecipitated with Hsp70 antibody as a positive control; N, protein lysates immunoprecipitated with control IgG as a negative control).
Hsp70 Downregulates the Expression of Several NFκB-Regulated Genes
Real-time polymerase chain reaction of four representative pro-inflammatory NFκB-regulated genes, TNF-α, IL-1β, ICAM-1, and iNOS was performed from brains exposed to tMCAO or LPS exposure. Our results show that while ischemic hemispheres from Wt mice have several-fold greater expression of TNF-α, ICAM-1, iNOS, and IL-β at 24 h after tMCAO compared with the corresponding contralateral hemispheres, the increased expression of these genes was significantly less among the Hsp Tg mice (Figure 9A). Ischemic hemispheres from the transgenic mice had lower expression of all four genes compared with Wt mice. Likewise, in the brain inflammation model, LPS induced several fold greater expression of these same genes in Wt mice that were downregulated in Hsp70 Tg mice (Figure 9B).
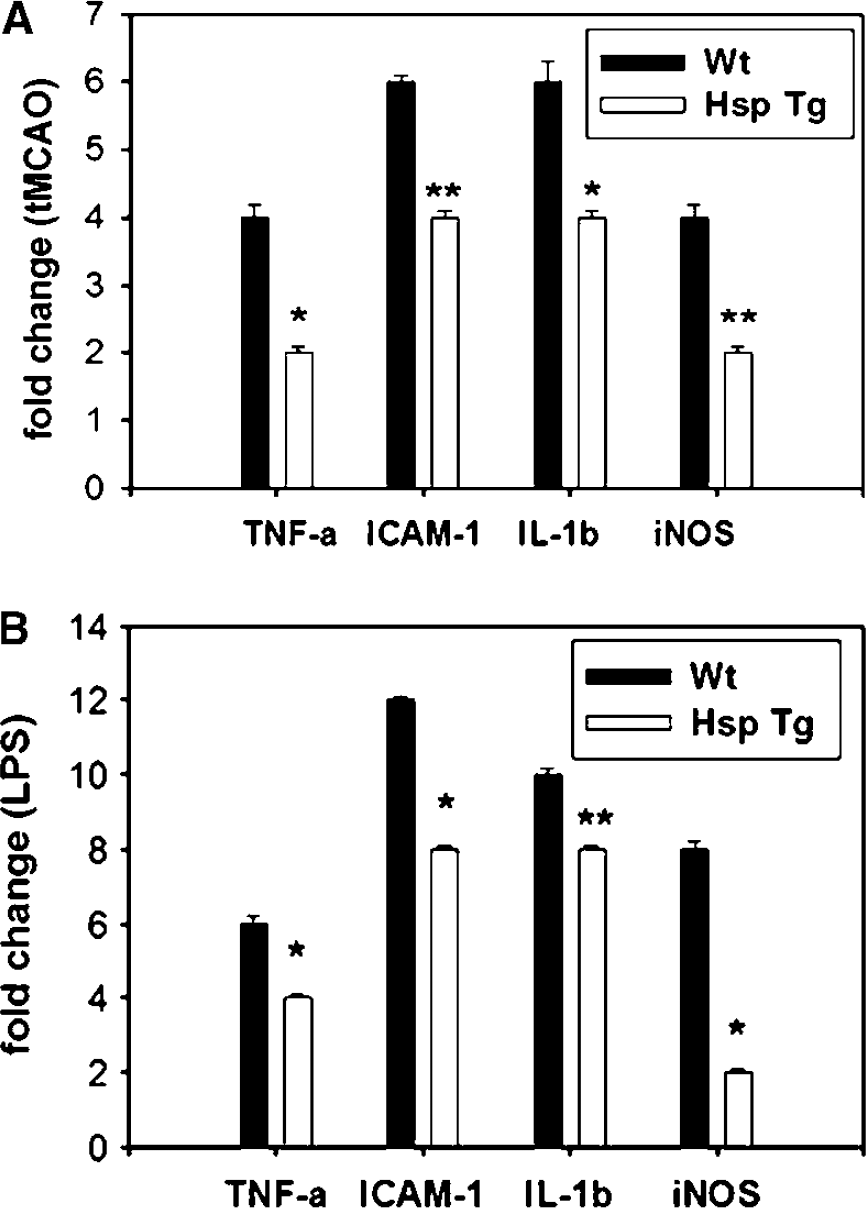
Down-regulation of expression of several representative NFκB-dependent pro-inflammatory genes in Hsp Tg mice. TaqMan Real-time PCR was used to estimate the expression of 4 different NFκB-dependent pro-inflammatory genes. Compared with Wt, expression of TNF-α, IL-1β, ICAM-1 and iNOS was significantly inhibited at the mRNA level among Hsp Tg mice following both brain ischemia (
Discussion
The cytotoxic properties of activated microglia after brain ischemia have been well documented (Block and Hong, 2005), and several lines of study have shown reduction in infarct size by inhibiting various inflammatory mediators (Zheng and Yenari, 2004). We show here that Hsp70 overexpression decreases microglia-induced cytotoxicity, microglial/macrophage activation and downregulates several pro-inflammatory genes. This work provides the first evidence of an anti-inflammatory property of Hsp70 to explain its neuroprotective function in brain ischemia. Further, we show that where microglia (an immune cell) potentiates ischemia-like injury to cultured astrocytes, Hsp70 completely prevents this. Moreover, the findings of this study show that Hsp70 blocks NF-κB activation and expression of several downstream inflammatory genes by interacting with the NF-κB:IκB complex and preventing the phosphorylation of IκB. To our knowledge, this work is the first of its kind.
Earlier work has shown that Hsp70 and other Hsps are capable of modulating inflammatory responses, but little is known as to whether this is the case in brain ischemia. Various studies have documented both pro- and anti-inflammatory properties of Hsp70. As a pro-inflammatory molecule, Hsp70 is thought to bind with receptors on antigen-presenting cells, stimulate cytokine secretion, upregulate co-stimulatory molecule expression (Basu et al, 2000; Basu and Srivastava, 2000), and facilitate antigen presentation (Moseley, 1998; Srivastava, 2002). As an anti-inflammatory molecule, Hsp70 decreases the release of inflammatory mediators in models of endotoxemia and cardiopulmonary bypass-induced inflammation (Hayashi et al, 2002; Klosterhalfen et al, 1996; Van Molle et al, 2002). Whether Hsp70 plays a role in inflammation due to brain ischemia has not been extensively studied. Earlier work has documented Hsp70 expression in microglia after experimental stroke (Soriano et al, 1994), and we have recently shown in our labs that MMP-9, one of several genes regulated by NF-κB, was reduced in cultured Hsp70 overexpressing astrocytes exposed to in vitro ischemia (Lee et al, 2004). Data presented here are consistent with Hsp70 as an anti-inflammatory molecule, regulating inflammation at the transcriptional level.
The mechanism of this anti-inflammatory effect may lie in Hsp70 interactions with NF-κB and its regulatory proteins. Nuclear factor-κB is an integral transcription factor mediating inflammatory responses to a variety of signals, and has been documented in cerebral ischemic injury (Schneider et al, 1999; Seegers et al, 2000). Nuclear factor-κB is normally sequestered in the cytoplasm where it is bound to a family of inhibitory proteins known as the inhibitor of NF-κB (IκB). Stimuli activate an upstream kinase (IκB kinase, IKK), which results in the phosphorylation of IκB leading to its ubiquitination and proteasomal degradation (Rothwarf and Karin, 1999). IκB degradation liberates NF-κB to enter the nucleus and induce gene expression. Anti-inflammatory properties of Hsp70 have been shown to be due to inhibition of NF-κB in models of heat stress and inflammation (Feinstein et al, 1996, 1997; Heneka et al, 2000). Glial cells exposed to heat stress (which causes induction of Hsp70) or transfected with Hsp70 followed by stimulation with LPS experienced less nuclear NF-κB translocation and less iNOS expression, an NFκB-regulated gene. Similarly, in a model of brain inflammation, heat stress was found to inhibit NF-κB activation, iNOS expression, and macrophage/microglial activation (Heneka et al, 2000). A recent study by Ran et al (2004) showed that Hsp70 can, in fact, directly interact with IKK-γ, and prevent IκB phosphorylation.
In this study, NF-κB activation was evaluated by using three different assays, and the data from these methods consistently show inhibitory effects of Hsp70 on NF-κB activation in both brain and microglia. Moreover, expression of some of NFκB-related inflammatory genes was significantly downregulated by Hsp70 overexpression. Furthermore, similar observations were made in two different models of brain inflammation owing to ischemic stroke and LPS exposure. The latter model is an important control, as inflammation in response to brain ischemia is proportional to the amount of injury. The LPS model would argue against the possibility that anti-inflammatory observations made in the ischemia model could be due to consequences of upstream factors, as we have previously shown that this model, while causing an inflammatory response, does not result in cell death (Deng et al, 2003).
Our data indicate that the mechanism of Hsp70 inhibitory effect on NF-κB activation appears to occur through the prevention of IκB phosphorylation. Our findings indicate that Hsp70 was found to associate with NF-κB p65 and IκBα, suggesting the possibility that Hsp70 physically binds to the NF-κB:I κBα complex, thereby stabilizing the complex and tethering NF-κB in the cytosol as a result. Our data are consistent with prior work in lymphoma cells, which showed that Hsp70 interacts with NF-κB and IκBα (Guzhova et al, 1997). We also found that Hsp70 does not interact with IKKγ. In contrast, Ran et al (2004) showed that Hsp70 interacts with IKKγ but not NF-κB, IκB, or other IKK. The reasons for these discrepancies are unclear, but may stem from the differences in the experimental systems used.
Although the present study links the neuro protective function of Hsp70 to an anti-inflammatory mechanism, this does not exclude other parallel mechanisms of protection. For example, two different mechanisms with respect to Hsp70 neuroprotection have recently been suggested that involve its ability to prevent protein aggregation (Giffard et al, 2004; Xu et al, 2006) and apoptosis (Matsumori et al, 2005; Tsuchiya et al, 2003). We therefore propose that an additional mechanism may lie in an anti-inflammatory effect regulated at the transcriptional level.
The major focus of the present work concentrated on the early inflammatory events that have been shown to contribute to the exacerbation of ischemic brain injury. However, it has also been suggested that inflammation is important in recovery and repair at later time points. We recognize that the delineation of Hsp70 anti-inflammatory functions at later time points merits further investigation, but is clearly beyond the scope of the current work. Nevertheless, the present work is the first to establish an anti-inflammatory property of Hsp70 and its interaction with NF-κB in the context of the brain ischemia. The findings not only provide a mechanistic basis for the neuroprotective effects of Hsp70 on brain ischemia, but would further support the potential of developing Hsp70 as a therapeutic against a variety of conditions, given that it may protect by several mechanisms.
Footnotes
Acknowledgements
We thank Yanli Qiao Nina Dunphy, Joyce Ma, and Yibing Ouyang for expert technical assistance and Dr Raymond A Swanson for providing the Hsp70 transgenic mice, and Beth Hoyte for help in preparing figures.