
Abstract
Nuclear factor-κB (NFκB) is a transcription factor that is activated after cerebral ischemia. NFκB activation leads to the expression of many inflammatory genes involved in the pathogenesis of stroke. The authors previously showed that mild hypothermia is protective even when cooling begins 2 h after stroke onset. In the present study, they examined the influence of hypothermia on NFκB activation. Rats underwent 2 h of transient middle cerebral artery occlusion. Brains were cooled to 33°C immediately after or 2 h after occlusion, and maintained for 2 h. After normothermic ischemia (brain temperature at 38°C), NFκB cytoplasmic expression, nuclear translocation, and binding activity were observed as early as 2 h in the ischemic hemisphere and persisted at 24 h. Hypothermia decreased NFκB translocation and binding activity but did not alter overall expression. Hypothermia also affected the levels of NFκB regulatory proteins by suppressing phosphorylation of NFκB's inhibitory protein (IκB-α) and IκB kinase (IKK-γ) and decreasing IKK activity, but did not alter overall IKK levels. Hypothermia suppressed the expression of two NFκB target genes: inducible nitric oxide synthase and TNF-α. These data suggest that the protective effect of hypothermia on cerebral injury is, in part, related to NFκB inhibition due to decreased activity of IKK.
Hypothermia has been shown both clinically (Bernard et al., 2002; Hypothermia After Cardiac Arrest Study Group, 2002) and experimentally (Clark et al., 1996; Colbourne et al., 1997; Han et al., 2002; Maier et al., 1998) to be neuroprotective. Proposed mechanisms of action of hypothermia include attenuation of blood–brain barrier permeability (Dietrich et al., 1990; Smith and Hall, 1996), excitatory amino acid accumulation (Busto et al., 1989), and free radical generation (Thoresen et al., 1997). Recently, there have been reports that hypothermia may attenuate the inflammatory response to cerebral ischemia (Chatzipanteli et al., 2000; Han et al., 2002; Inamasu et al., 2000, 2001; Maier et al., 1998; Sutcliffe et al., 2001; Toyoda et al., 1996; Wang et al., 2002). Inflammation plays an important role in the pathogenesis of cerebral ischemia and secondary damage, and antiinflammatory therapies for stroke have been the subject of intense investigation (Barone and Feuerstein, 1999).
Nuclear factor-κB (NFκB) is one of the most important transcription factors playing a pivotal role in mediating inflammatory responses to a variety of signals, including inflammatory cytokines, phorbol esters, growth factors, bacterial and viral products, oxidative stress, hypoxia-reoxygenation, and ultraviolet light and irradiation (Baeuerle and Henkel, 1994). NFκB is present in the cytoplasm and serves as a critical regulator of the inducible expression of genes involved in immunity, inflammation, and cell adhesion, as well as in cell growth and death (Ghosh et al., 1998; May and Ghosh, 1998; Verma and Stevenson, 1997). NFκB is also involved in acute-phase and inflammatory responses that potentiate ischemic injury (Clemens et al., 1997; Schneider et al., 1999; Seegers et al., 2000), activating many genes involved in the pathogenesis of cerebral ischemia, such as inducible nitric oxide synthase (iNOS), interleukin-1β, tumor necrosis factor (TNF)-α, intercellular cell adhesion molecule (ICAM)-1, cyclooxygenase-2, and interleukin-6 (Smith and Hall, 1996). NFκB is normally sequestered in the cytoplasm where it is bound to a family of inhibitory proteins known as NFκB inhibitors (IκB). The IκB family includes IκB-α, IκB-β, IκB-γ, and IκB-ε. A major IκB and the one most often studied is IκB-α. Stimuli activate an upstream kinase (IκB kinase, IKK), which results in the phosphorylation of IκB and leads to its ubiquitination and proteasomal degradation (Rothwarf and Karin, 1999). IκB degradation liberates NFκB, which enters the nucleus and induces gene expression. This process is eventually terminated through NFκB-induced synthesis of IκB followed by cytoplasmic resequestration.
IKK contains two catalytic subunits, IKK-α and -β, both of which have kinase domains. IKK-γ, in conjunction with IKK-α and IKK-β, is a component of the IKK complex (Rothwarf and Karin, 1999) and facilitates IKK activity. Recent data describe a canonical activation pathway that depends on the phosphorylation of IκB by IKK-β, whereas IKK-α is responsible for specialized activities via NFKB2 and p100 processing (Senftleben and Karin, 2002).
We and others previously showed that mild hypothermia is associated with decreases in several NFκB-regulated proteins such as iNOS (Han et al., 2002), ICAM-1 (Inamasu et al., 2001; Wang et al., 2002), and inflammatory cytokines (Goss et al., 1995). Although it is established that inflammation contributes to cerebral ischemic injury and that mild hypothermia is an effective neuroprotectant, it is not known whether NFκB is altered to achieve hypothermic protection.
MATERIALS AND METHODS
Experiments were carried out according to the guidelines for the care and use of laboratory animals approved by the Stanford University Administrative Panel on Laboratory Animal Care. Animals were housed under diurnal lighting conditions in a temperature-controlled environment and with free access to food and water until the day of the experiment.
Focal cerebral ischemia
Male Sprague-Dawley rats weighing between 290 and 320 g were anesthetized with isoflurane during surgical procedures. A femoral artery was cannulated for the continuous monitoring of arterial blood pressure and blood sampling. Physiologic parameters were monitored and maintained in the normal range as shown previously (Han et al., 2002). Blood gases were measured with an automatic pH/Blood Gas Analyzer Model 178 (Ciba Corning Diagnostics Corp., Medfield, MA, U.S.A.). Ischemia was induced using an occluding intraluminal suture. An uncoated 30-mm segment of 3–0 nylon monofilament suture with a flame-rounded tip was inserted into the stump of the common carotid artery and advanced into the internal carotid artery approximately 19 or 20 mm from the bifurcation, to occlude the ostium of middle cerebral artery (MCAO). At the end of the ischemic period, the suture was removed and the animal was allowed to recover. Sham-operated animals were treated in the same manner as the ischemic animals, but no ischemia was applied. During surgery, rectal temperature was maintained between 37°C and 38°C corresponding to a brain temperature of 38°C to 39°C (Yenari et al., 2000). Mild hypothermia (33°C rectal temperature corresponding to an ischemic brain temperature of 33°C and a nonischemic brain temperature of 35°C) was induced as previously described using a method that consistently produces neuroprotection (Han et al., 2002; Maier et al., 1998; Yenari et al., 2000). During surgery, the rectal temperature was maintained between 37°C and 38°C, corresponding to a brain temperature of 38°C to 39°C (Yenari et al., 2000). Mild hypothermia (33°C rectal temperature) was achieved as described previously (Maier et al., 1998; Yenari et al., 2000). In brief, total body cooling was achieved by spraying alcohol onto the animal and cooling it to the desired temperature with a fan. Rewarming was achieved using a heating pad, which was placed under the animal, and a lamp was positioned over the animal's body. At the end of the experiment all animals were returned to normothermia. Both cooling and rewarming phases each required approximately 10 minutes to achieve the target temperature in the hypothermic groups. Cooling began at the onset of ischemia (intraischemic hypothermia) or was delayed by 2 h (delayed hypothermia; cooling began immediately after the suture was removed) and was maintained for 2 h. The animals were killed by a carbon dioxide overdose 2, 6, 12, and 24 h after MCAO onset and prepared for further analysis.
Tissue preparation
Animals were perfused with normal saline, and brains were removed and sectioned into four coronal slices containing areas of maximal ischemic damage (from 4-mm anterior to bregma to 8-mm posterior to bregma). Each slice was dissected into four parts: ischemic cortex, ischemic subcortex, contralateral cortex, and contralateral subcortex. The tissues were weighed and processed for microwell NFκB assay, Western blots, IKK assay, and RNA isolation. All assays were performed in triplicate.
Immunohistochemistry
Immunohistochemistry was performed to identify the cells expressing NFκB. Six-micrometer-thick sections were cut from paraffin-embedded brains. After deparaffinization, sections were treated for endogenous peroxidases with 0.03% H2O2 and blocked in 1% bovine serum albumin and 5% normal goat serum, then incubated with primary antibody against NFκB p65 (1:200, sc-109; Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.), followed by the anti-rabbit immunoglobulin G (IgG) secondary antibody (1:200, BA-1000; Vector Labs, Burlingame, CA, U.S.A.). Antibodies were detected using the Vector ABC kit (Elite Vectastain ABC kit; Vector Labs) and colorized with 0.05% diaminobenzidine (DAB; Vector Labs). Negative controls were run in parallel using adjacent sections incubated with IgG instead of the primary antibody. Hematoxylin and eosin staining was also performed on adjacent sections.
SDS-PAGE and immunoblotting
Brain samples were homogenized in Laemmli's lysis buffer plus protease inhibitors. Aliquots containing 25 μg protein were subjected to 10% SDS-PAGE. Protein bands were transferred to polyvinylidene diflouride membrane (IPVH00010; Millipore, Bedford, MA, U.S.A.) and probed by incubating in the primary antibodies, followed by a horseradish peroxidase–conjugated secondary antibody (1:5,000, sc-1004 or sc-1005; Santa Cruz Biotechnology). We used the following primary antibodies: anti-NFκB p65 (1:1,000, sc-109; Santa Cruz Biotechnology), anti-IκB-α (1:500, 40903; Active Motif, Carlsbad, CA, U.S.A.), antiphosphorylated IκB-α (1:500, 40904; Active Motif), anti–IKK-α (1:500, SA-301; Biomol, Plymouth Meeting, PA, U.S.A.), anti–IKK-β (1:500; SA-302; Biomol), and anti–IKK-γ (1:500, 40908; Active Motif). To determine the specificity of the primary antibodies, we used antibodies preabsorbed with blocking peptides instead of primary antibodies. Blots were visualized using the enhanced chemiluminescence system (Amersham, Piscataway, NJ, U.S.A.) according to the manufacturer's directions, and exposed to x-ray film. Equal protein loading was confirmed by measuring β-actin (1:5,000, anti-β-actin, A5441; Sigma, St. Louis, MO, U.S.A.). Densitometric measurements were made from the film using a GS-700 imaging densitometer (Bio-Rad, Hercules, CA, U.S.A.), then quantified using Multi-Analyst (Bio-Rad). For quantification of relative protein expression, the optical density of the protein band of interest was normalized to the optical density of β-actin on the same gel.
Microwell colorimetric nuclear factor-κB assay
The binding ability of NFκB to DNA consensus sequences was measured using a commercially available kit (Trans-AMTM NFκB p65, 40096; Active Motif). This method was used as an alternative to electrophoretic mobility shift assay (EMSA) and has been reported to be more sensitive and quantitative than EMSA (Shen et al., 2002). In this assay, tissue lysates were tested for their ability to bind to a double-stranded oligonucleotide probe containing the consensus binding sequence for NFκB. We followed the manufacturer's protocol, which was modified from that of Renard et al. (2001). Samples were homogenized in 3 mL ice-cold lysis buffer (20 mmol HEPES, pH 7.5; 350 mmol NaCl; 20% glycerol; 1% Igepal-CA630; 1 mmol MgCl2; 0.5 mmol EDTA; 0.1 mmol EGTA) per gram tissue. Lysates were centrifuged at 10,000g for 10 minutes at 4°C. The supernatant was used to measure the protein content by a Bradford-based assay (Bio-Rad). NFκB activity was determined by the sample's ability to bind to consensus sequences (5′-GGGACTTTCC-3′) in a 96-well plate. A primary antibody that recognizes an epitope on p65 and is accessible only when NFκB is activated and bound to its target DNA was added to the wells, followed by a secondary horseradish peroxidase–conjugated antibody. Developing solution (tetramethylbenzidine) was added and the colorimetric reaction was stopped by adding stop solution (0.5-mol/L H2SO4). After stopping the reaction, absorbance was measured on a spectrophotometer within 5 minutes at 450 nm with a reference wavelength of 655 nm. HeLa whole-cell extract was used as positive control for NFκB activation. The NFκB wild-type and mutated consensus oligonucleotides were used in order to monitor the specificity of the assay. A wild-type oligonucleotide should compete with NFκB for binding to the probe immobilized on the plate, but the mutated consensus oligonucleotide should have no effect on NFκB binding.
IKK activity assay
IKK activity was determined using a protocol modified from Li et al. (2001). This assay is based on the amount of phosphorylated IkB-α synthesized by IKK, reflecting its activity. It is equivalent to the amount of radioactive phosphate incorporated to the IkB-α substrate. Fresh frozen brain sections from the periinfarct area were homogenized in lysis buffer (1 mL of buffer per 100 mg of tissue, 50 mmol HEPES, pH 7.3; 250 mmol NaCl; 1 mmol ethylenediamine tetraacetic acid [EDTA]; 1% Igepal; protease inhibitor cocktail, p8340, Sigma; 1 mmol sodium orthovanadate; 50 mmol sodium fluoride) using a dounce homogenizer at 4°C. The lysates were transferred to microcentrifuge tubes and centrifuged at 10,000g for 10 minutes at 4°C. Supernatants were collected and the protein content was measured (BioRad). Sample protein (100 μg) was immunoprecipitated with 1 μg IKK antibody at 4°C for 2 h (anti–IKK-β sc-7607, anti–IKK-γ sc-8330; Santa Cruz Biotechnology). Twenty microliters of Protein A/G PLUS-Agarose-conjugated beads (sc-2003; Santa Cruz Biotechnology) was added and incubated overnight at 4°C. The precipitates were collected by centrifugation at 2,500 rpm for 5 minutes at 4°C. The beads were then washed twice with lysis buffer, once with kinase buffer (10 mmol HEPES, pH 7.3; 1 mmol MnCl2; 5 mmol MgCl2; 12.5 mmol β-glycero 2-phosphate; 100 μmol sodium orthovanadate; 2 mmol sodium fluoride; 1 mmol dithiothreitol). The precipitated IKK were reconstituted with 80 μL kinase assay buffer and 20 μL ATP solution (2 mmol ATP, 0.925 μg glutathione S-transferase-IκB-α substrate, sc-4094; Santa Cruz Biotechnology; and 5 μCi γ-32-P ATP) was added. The reaction mixture was incubated for 30 minutes at room temperature and 30 μL stop solution (200 mmol EDTA, 200 mmol ATP in 10 mmol HEPES) was added. The reaction solutions were blotted onto the phosphocellulose membranes (Whatman P81) to select phosphorylated proteins specifically, washed with water for 1.5 minutes (twice) and then with ethanol for 1 minute. Each membrane was put into a scintillation vial and the radioactivity was counted using a γ-counter. Counts per minute were measured, which reflects the amount of radioactive phosphate and thus IKK activity.
RNA isolation and reverse transcription–polymerase chain reaction assay
Total RNA was isolated from the brain sections (RNeasy Midi kit, #75144; Qiagen, Valencia, CA, U.S.A.). The concentration of total RNA isolated was quantified by ultraviolet spectrophotometry at 260/280 nm. Reverse transcription–polymerase chain reaction (RT-PCR) assays were performed with ThermoScript RT-PCR system (11146–016; Invitrogen, Carlsbad, CA, U.S.A.). One microgram of total RNA from each sample was reverse-transcribed into complementary DNA (cDNA), and PCR was performed following the manufacturer's protocol. The following primers were used: TNF-α32, sense: 5′ GCTGCCCCGACTATGTGCTCCTCA 3′, antisense: 5′ GACGCCCCGGCCTTCCAA-ATAAAT 3′; GAPDH32, sense: 5′ TGAAGGTCGGTGTGAACGGATTTGG 3′, antisense: 5′ ACGACATACTCAGCACCAGCATCAC 3′; iNOS33, sense: 5′ CTGCATGGAAC-AGTATAAGGCAAAC 3′ antisense: 5′ CAGACAGTTTCTGGTCGATGTCATGA 3′. Amplification cycles were performed in a thermal cycler (Techne Inc., Princeton, NJ, U.S.A.). Before the cycle started, the sample was preheated at 94°C for 2 minutes. Each cycle consisted of 35 cycles of 45 seconds at 94°C, 30 seconds at 63°C, 45 seconds at 72°C (TNF-α), 35 cycles of 45 seconds at 94°C, 30 seconds at 55°C, 45 seconds at 72°C (GAPDH), 40 cycles of 35 seconds at 93°C, 45 seconds at 63°C, and 45 seconds at 72°C (iNOS) followed by 10 minutes at 72°C. Negative control reactions were performed with each batch of cDNA synthesis without reverse transcriptase. The PCR products were analyzed by electrophoresis on 1% agarose gels (Agarose; Gibco BRL, Carlsbad, CA, U.S.A.) with 1× tris/borate/EDTA buffer (Gibco BRL). The gels were stained with ethidium bromide and densitometric measurements were made from the film using a GS-700 imaging densitometer (Bio-Rad) and then quantified using Multi-Analyst (Bio-Rad). The messenger RNA (mRNA) levels of constitutively expressed GAPDH were determined to control for differences in cDNA synthesis efficiency. Thus, cDNA levels were calculated by comparison with synthesis of GAPDH. To account for slight differences in RNA sample loading, all values obtained with the TNF-α and iNOS were normalized to the values obtained with the GAPDH.
Statistical analysis
Data are presented as means ± SEM. Comparisons between groups were performed with Sigmastat software (SPSS, Chicago, IL, U.S.A.) using standard statistical methods and were analyzed by one-way ANOVA followed by pairwise multiple comparison procedures. Statistical significance was determined at the P < 0.05 level.
RESULTS
Ischemic injury and nuclear factor-κB immunohistochemistry
The hematoxylin–eosin stains showed the morphologic characteristics consistent with ischemic cell injury in the territory of the ipsilateral middle cerebral artery (Fig. 1B). Cellular injury was less severe in hypothermia-treated brains (Fig. 1C), with fewer shrunken, pyknotic-appearing cells compared with normothermic brains (Fig. 1B). Such morphologic changes were not observed in sham-operated brains (Fig. 1A). These data are consistent with the robust protection we have previously reported with hypothermia (Han et al., 2002). Cytoplasmic and nuclear staining of the p65 subunit was barely detectable in uninjured brains (Fig. 1D), but a variety of different cell types (identified by morphological characteristics) showed increased NFκB staining in both the cytoplasm and nuclei of ischemic hemispheres. NFkB staining was present in all cell types including neurons as early as 2 h and as late as 1 d after the onset of ischemia. Ischemia induced NFκB expression, but normothermic brains frequently showed increased nuclear staining (Fig. 1E), whereas staining in hypothermic brains was largely confined to the cytoplasm (Fig. 1F). In the contralateral hemisphere, NFκB-positive cells were observed, although the number was much smaller than in the ischemic hemisphere (data not shown)
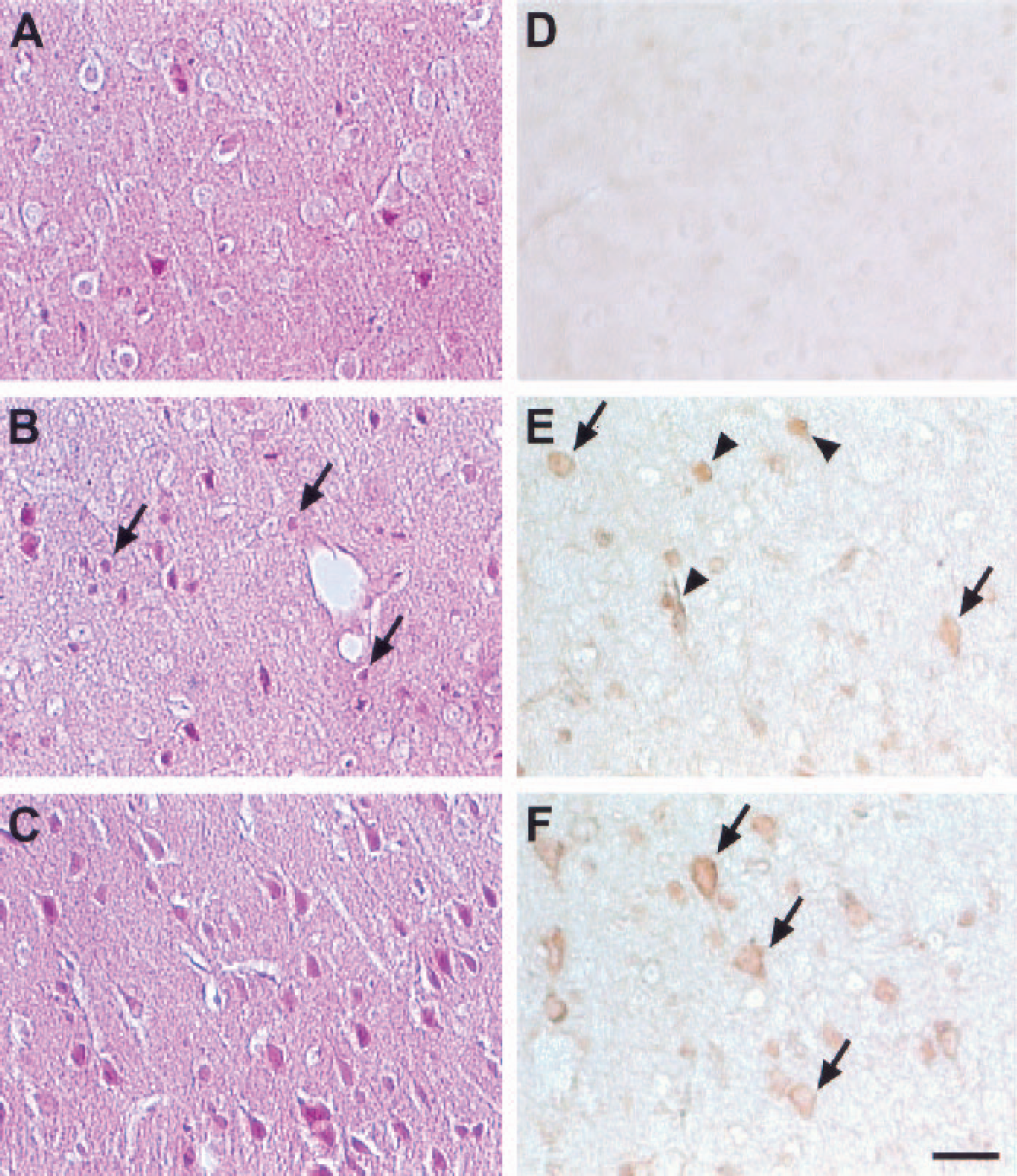
Mild hypothermia reduced the extent of ischemic cell injury
Nuclear factor-κB binding activity
Activation of NFκB is due to increased DNA binding of NFκB after its release from IκB. Since the p65 subunit, which has potent transcriptional activation domains, is known to be important in ischemia (Rothwarf and Karin, 1999; Schneider et al., 1999; Seegers et al., 2000), we used a p65-specific antibody to assess DNA binding with microwell colorimetric assay. The NFκB wild-type and mutated consensus oligonucleotides were used to monitor the specificity of the assay. Wild-type oligonucleotide competed for NFκB binding to the probe immobilized on the plate, but the mutated consensus oligonucleotide had no effect on NFκB binding (data not shown). After 2 h of MCAO, specific NFκB binding activity was increased in normothermic ischemic brains, an effect observed also at 24 h. When mild hypothermia was applied either during or after ischemia, binding activity was significantly decreased (Fig. 2).
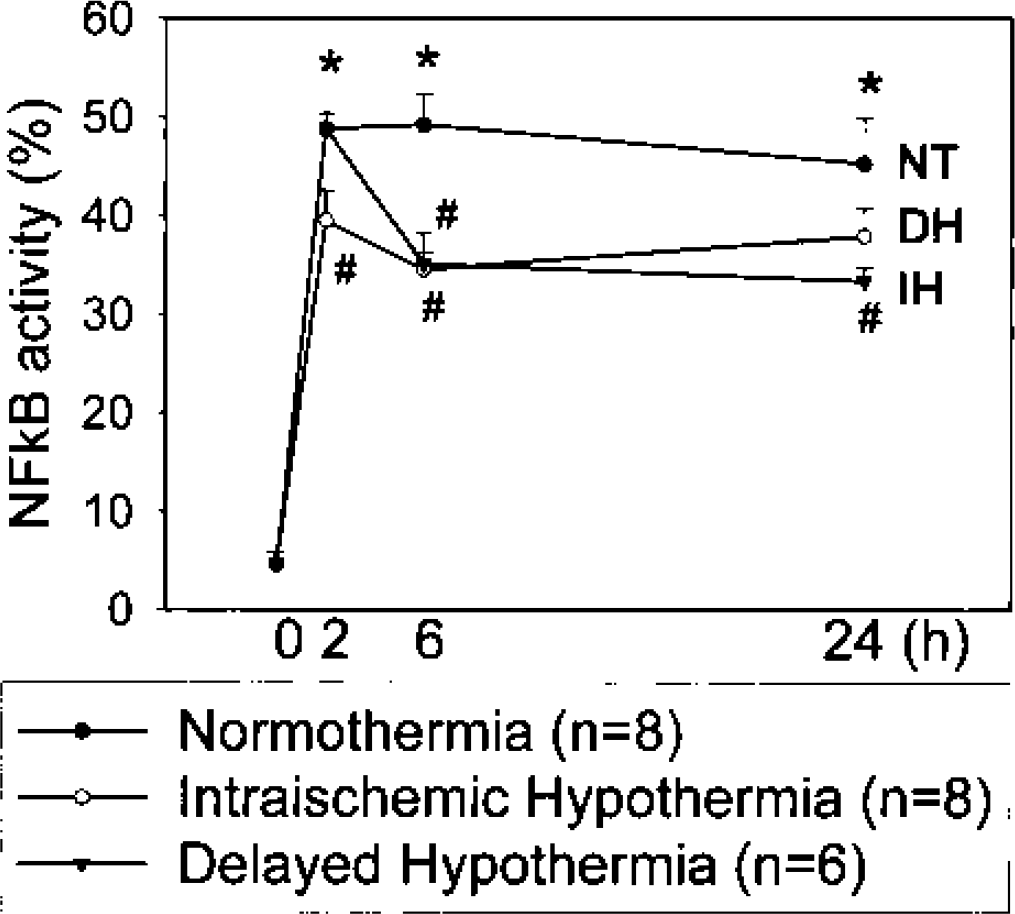
Cerebral ischemia increased DNA binding of nuclear factor-κB (NFκB) p65, and mild hypothermia attenuated this increase. Rats were subjected to 2 h of middle cerebral artery occlusion (MCAO), and brains were taken 0, 2, 6, and 24 h after MCAO onset. At each time point, periinfarct areas were harvested and prepared for NFκB-binding activity assay. Tissue samples from sham-operated rats were taken from the same regions as those from ischemic brains and served as controls. After ischemia NFκB-binding activity increased as early as 2 h and was maintained at 24 h after MCAO. Mild hypothermia applied during or delayed after ischemia suppressed binding activity significantly. *P < 0.05 normothermia vs. sham; #P < 0.05 hypothermia vs. normothermia (ANOVA followed by Tukey test).
Western blots of nuclear factor-κB, IκB-α, and phosphorylated IκB-α
Quantitative measurement of NFκB, IκB-α, and phosphorylated IκB-α (p-IκB-α) proteins prepared from periinfarct areas of the ischemic brains 2, 6, 24, and 72 h after MCAO was performed using Western blot analysis to examine the regulatory alterations in the NFκB pathway in ischemic brains under conditions of normothermia and hypothermia. NFκB is composed of two subunits, both of which belong to the Rel family of proteins. The most frequently found NFκB heterodimer is p65-p50. In quiescent cells, most of the p65-p50 heterodimers are retained in cytoplasm as a complex with IκB. We detected NFκB p65 protein bands in sham-operated animal brains. The amount of NFκB p65 increased as early as 2 h after ischemia, peaked at 6 h, and persisted at 24 h. Seventy-two hours after MCAO, the amount of NFκB p65 decreased to baseline levels (197.2% of control at 2 h, 303.2% at 6 h, 263.9% at 24 h, and 151% at 72 h). NFκB p50 showed similar expression patterns (Fig. 3A). Hypothermia decreased overall NFκB p65 levels at 6 and 24 h compared with normothermia (Fig. 3B).
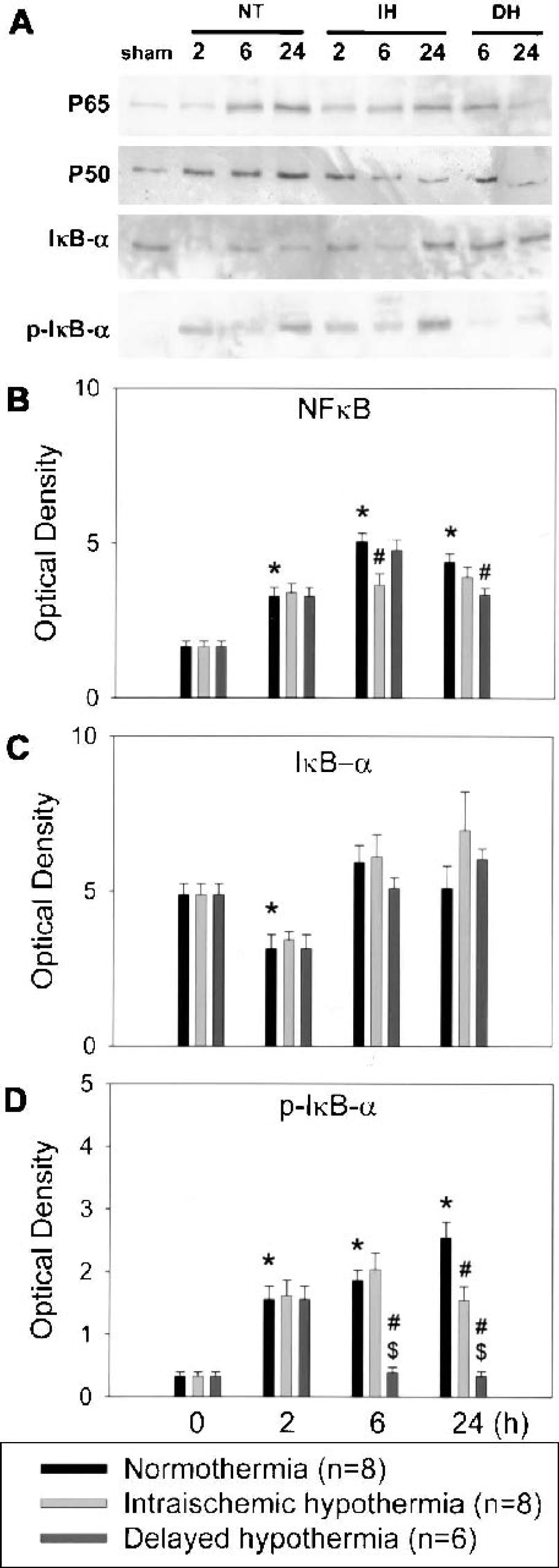
Representative Western blots using antibodies against nuclear factor-κB (NFκB) p65 (p65), NFκB p50 (p50), IκB-α, and phosphorylated IκB-α (p-IκB-α) are shown
To examine alterations in NFκB regulation by its inhibitor protein IκB, we measured the amount of IκB-α, a major IκB family member. IκB-α was detected in sham-operated brains and decreased 2 h after MCAO and then increased thereafter. By 72 h, IκB-α returned to basal levels (64.2% of control at 2 h, 121.6% at 6 h, 104.5% at 24 h, and 99.1% at 72 h). Hypothermia had no significant effect on overall IκB-α expression (Fig. 3C). To examine the effect of hypothermia on IκB-α phosphorylation and degradation, we assessed the level of p-IκB-α and found that this phosphorylated protein increased after ischemia and reperfusion but was reduced by mild hypothermia, especially when cooling was delayed (Fig. 3D).
Western blots of IKK
We performed Western blots of individual IKK subunits using specific antibodies for α (involved in adaptive B-cell–mediated immunity), β (involved in activation of innate immunity and inflammatory responses), and γ (involved in regulation of the IKK complex). IKK-γ expression increased after ischemia, but these increases were attenuated by mild hypothermia. Furthermore, delayed hypothermia significantly suppressed overall IKK-γ levels compared with intraischemic hypothermia (Fig. 4B). Hypothermia did not significantly change ischemia-induced expression of IKK-α and -β subunits (Fig. 4A).
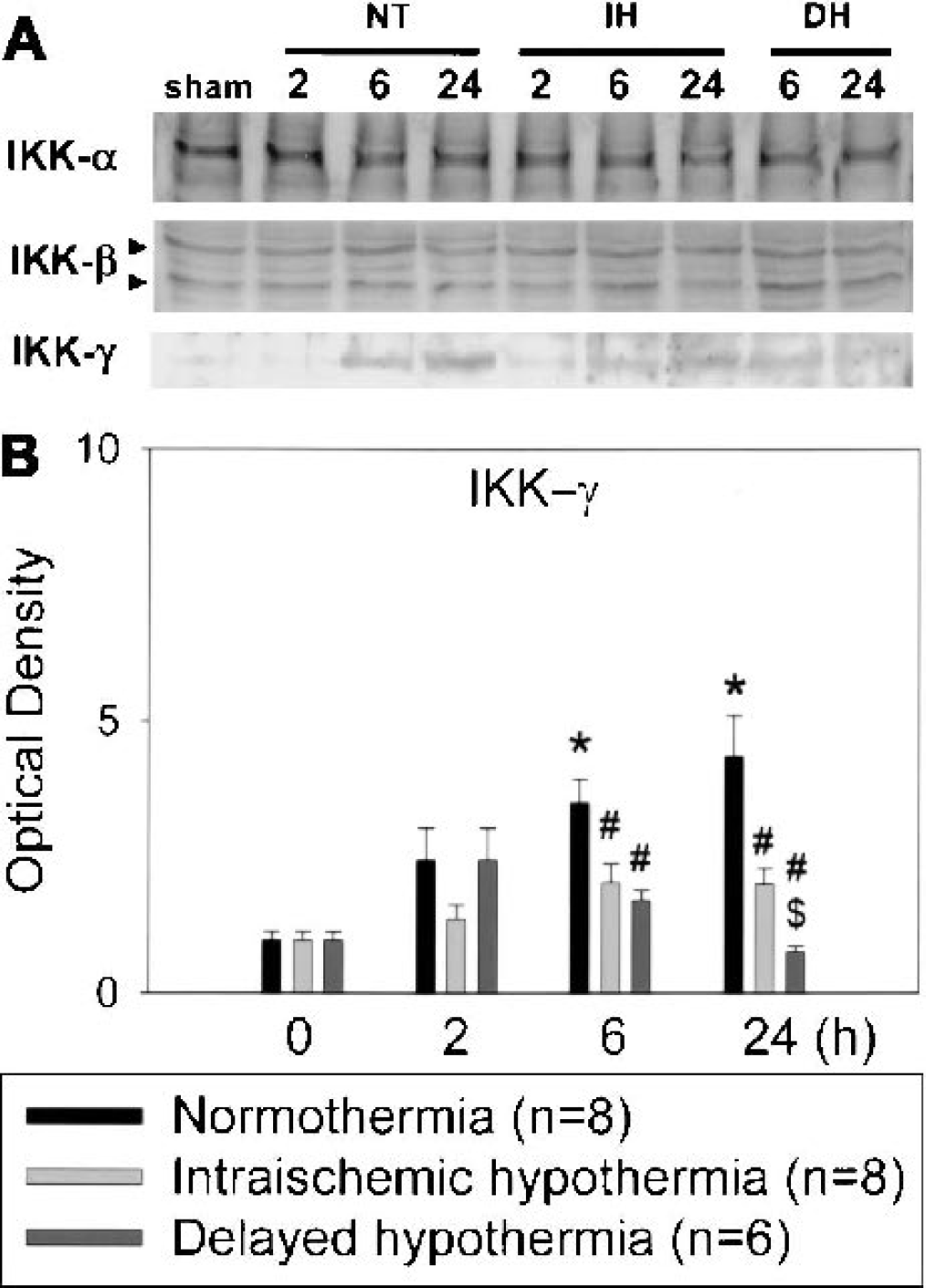
Representative Western blots using antibodies against IKK-α, -β, and -γ are shown
IKK activity assay
Since Western blot analysis indicated that mild hypothermia decreased expression of IKK-γ, we then determined whether enzymatic activity might also be altered by temperature. To examine alterations in the NFκB-activating pathway in ischemic brains that lead to increased phosphorylation and degradation of IκB-α, we initially assessed the activity of IKK complex. We selected IKK-β because it was known that IKK-β makes essential contributions to phosphorylation of IκB and plays important roles in inflammation-related gene transcriptions, whereas IKK-α is not essential (Senftleben and Karin, 2002). To evaluate any alterations in the activity of this enzyme complex, we immunoprecipitated the IKK complex using specific antibodies for IKK-β and -γ. The kinase assay performed on IKK immunoprecipitates indicated that IKK activity increased after ischemia and was significantly suppressed under both hypothermic conditions, but delayed hypothermia suppressed IKK-β activity more than intraischemic hypothermia (Fig. 5). The inhibition by hypothermia of IKK activity is consistent with our observation that phosphorylated IκB-α increases under normothermic ischemia compared with hypothermia.
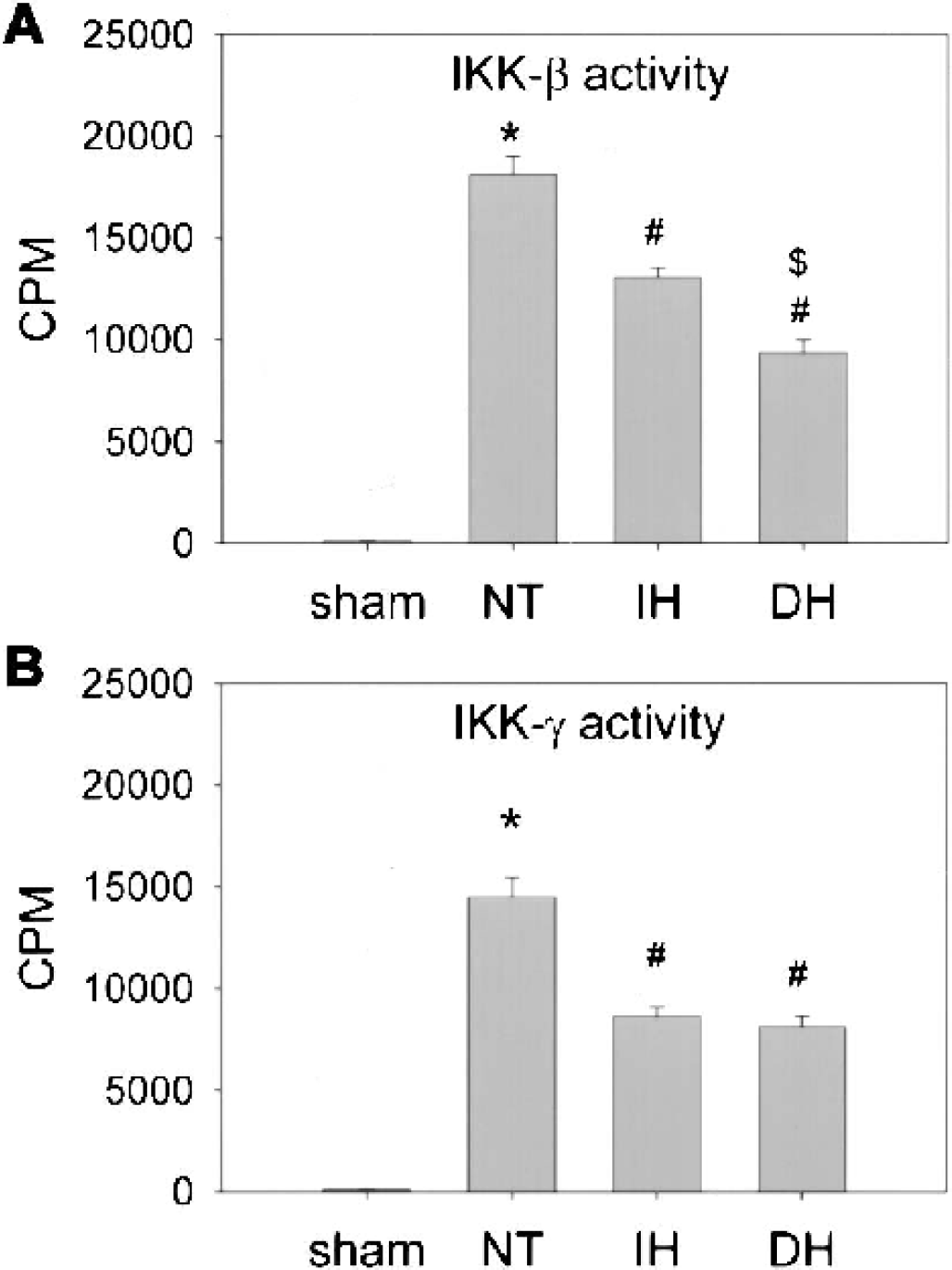
Hypothermia decreased IKK activity. IKK activity was analyzed by immunoprecipitation with anti–IKK-β or γ antibodies followed by addition of glutathione S-transferase (GST)-IkB-α substrate. Tissue samples were taken from the periinfarct regions of the ischemic brains. Mild hypothermia attenuated the activity of IKK-β 24 h after middle cerebral artery occlusion. Delayed hypothermia (DH) caused significant suppression of activity compared with intraischemic hypothermia (IH;
Gene expression
Because iNOS and TNF-α are NFκB-regulated genes, we examined whether hypothermia prevented NFκB target gene expression. mRNA levels in the periinfarct areas of the ischemic brains at 2, 6, 12, 24 h after MCAO were analyzed by RT-PCR. As shown in Fig. 6B, TNF-α mRNA levels increased after ischemia, and hypothermia significantly prevented the ischemic stimulation of TNF-α mRNA expression. Similarly, ischemia induced the expression of iNOS mRNA, which was decreased by hypothermia (Fig. 6C).
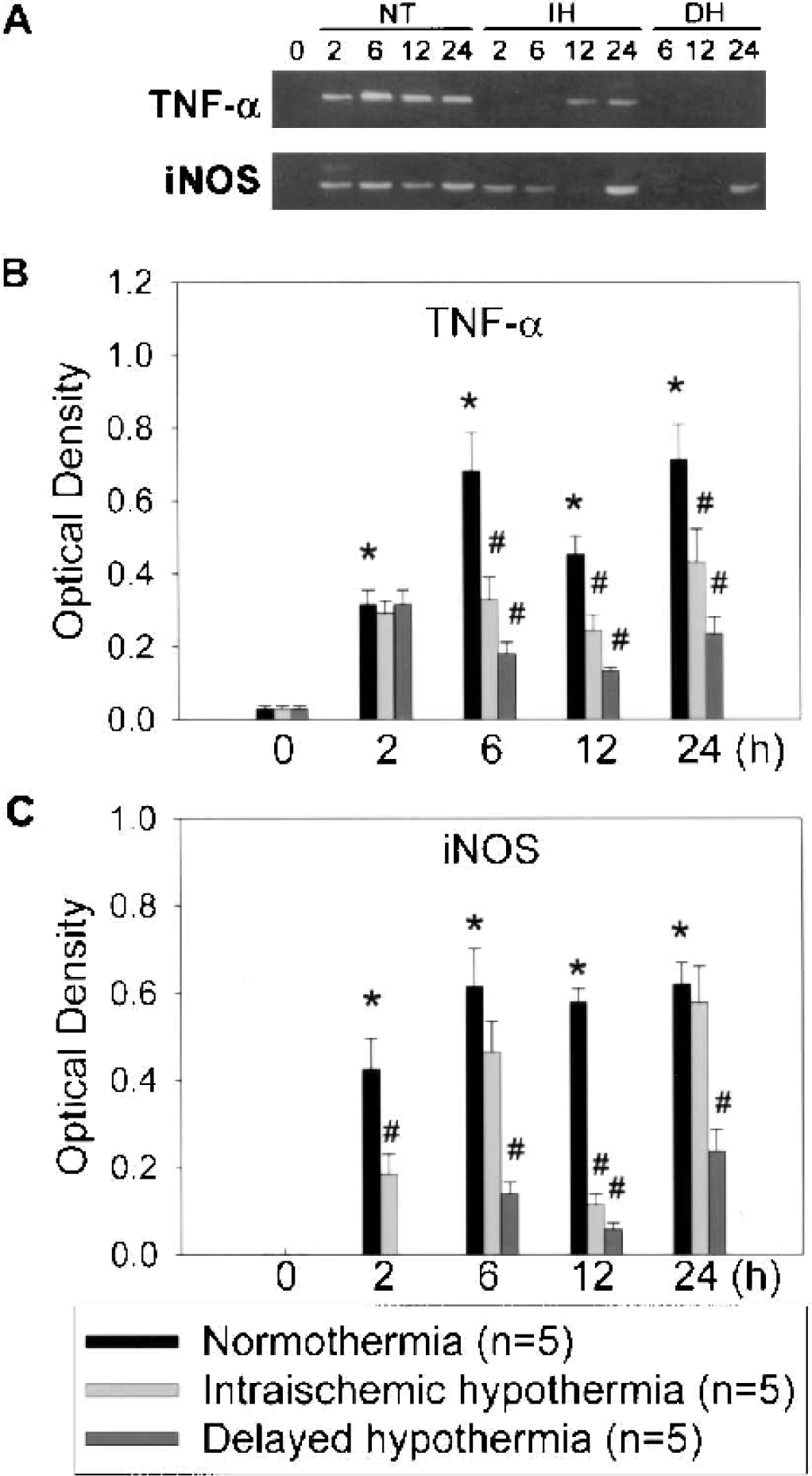
Mild hypothermia attenuated gene expression of inducible nitric oxide synthase (iNOS) and tumor necrosis factor-α (TNF-α) in the ischemic brains. Messenger RNA isolated from periinfarct areas at 0, 2, 6, 12, and 24 h after middle cerebral artery occlusion under conditions of normothermia (NT), intraischemic hypothermia (IH), and delayed hypothermia (DH) were subjected to reverse transcription–polymerase chain reaction with iNOS or TNF-α specific primer sets. Ischemic insults induced the expression of TNF-α and iNOS under conditions of NT. IH and DH suppressed iNOS and TNF-α expression. Representative gel electrophoresis of polymerase chain reaction products
DISCUSSION
We show here that mild hypothermia can suppress the inflammatory response by interfering with NFκB regulation at the level of its IKK. According to our present understanding of NFκB, its activation in cerebral ischemia is a multistep process. We systematically tracked various aspects of NFκB regulation in our stroke model. Mild hypothermia suppressed binding of NFκB to its consensus DNA sequences by decreasing IKK-γ expression and IKK enzymatic activity. This in turn led to reduced phosphorylation of IκB-α and nuclear translocation of NFκB. Ischemia-induced upregulation of the NFκB system was attenuated by mild hypothermia, and delayed hypothermia inhibited some of these actions more effectively than intraischemic hypothermia.
We found increased NFκB and translocation from cytoplasm to nuclei, along with increased NFκB binding to the corresponding DNA sequences, as early as 2 h after MCAO with persistent activity at 24 h. Overall NFκB levels were increased after ischemia and reperfusion, and this may be caused either by enhanced synthesis of NFκB itself or by decreased degradation. Since p50 or p52 binding to promoter sites does not stimulate transcription, both p50 and p52 lack an activating domain, whereas p65 has an activating domain and thus can act as a transcription enhancer. Differential translocation of different NFκB dimers can act as repressors or enhancers. In our experimental model, the amount of NFκB p50 increased along with p65, and this increase contributed to the NFκB transcriptional activity. IκB-α levels decreased immediately after ischemia but returned to basal levels during reperfusion, whereas the amount of phosphorylated IκB-α was significantly elevated in ischemic brains, indicating increased IKK activity. However, whereas hypothermia decreased phosphorylated IκB-α levels, total IκB-α levels were not changed. Since NFκB also stimulates IκB synthesis, the lack of changes in IκB levels may be explained by newly synthesized IκB. Although hypothermia showed a trend towards increasing IκB-α, this might have been confounded by increased IκB expression in normothermic brain resulting from greater NFκB action. Whether hypothermia itself necessarily alters IκB expression at the transcriptional or translational levels cannot be determined from the data presented here. Nevertheless, phosphorylated IκB-α was increased in normothermia, suggesting that more free NFκB was available to translocate to the nucleus, and this was attenuated by hypothermia.
Whereas the total amount of IKK-α and -β protein was unchanged, IKK-γ increased after ischemia (Fig. 4). Ischemia induced IKK activation involving both IKK-β and -γ subunits. However, hypothermia suppressed the IKK activity. IKK-γ has no catalytic domain itself; it modulates the activity of the IKK complex (Senftleben and Karin, 2002) by facilitating the association of both the IKK complex and IκB to increase IκB phosphorylation (Yamamoto et al., 2001). Hypothermic suppression of IKK activity may play a key role in attenuated phosphorylation of IκB-α, whereas IKK-γ may act as a modulator.
Transcription of two representative inflammatory genes regulated by NFκB, iNOS and TNF-α, was upregulated after ischemia. Hypothermia decreased transcriptional activity of NFκB, as indicated by mRNA levels of TNF-α and iNOS. This is consistent with our prior observations that hypothermia suppresses iNOS expression, particularly in inflammatory cells. Hypothermic suppression of iNOS expression was observed, but intraischemic hypothermia at 6 and 24 h did not suppress iNOS expression significantly, even though we observed significant suppression of NFκB activity under the same conditions. Thus, the general trends of iNOS and TNF-α gene expression are similar but not identical. Ginis et al. (2002) showed that different genes were regulated by different coactivator or adaptor proteins even though the genes were activated by the same transcription factor. Whereas this observation may explain the discrepancy of mRNA levels between iNOS and TNF-α, it is also possible that other transcription factors may be involved in regulating iNOS (Ricote et al., 1998) or TNF-α expression.
The observation of increased NFκB staining in a variety of different cell types after ischemia suggests that hypothermic attenuation of NFκB activity may not occur exclusively in inflammatory cells. Thus, hypothermic suppression of inflammation can be explained in two ways. First, hypothermia attenuates NFκB activity of the inflammatory cells, leading to direct inhibition of inflammation. Second, other cells that are protected by hypothermia (such as neurons) may have influenced surrounding cells less owing to decreased generation of inflammatory signaling proteins, and will indirectly decrease secondary stimuli to the inflammatory cells.
Interestingly, we found that delayed hypothermia resulted in more suppression of IκB phosphorylation, IKK expression and activity, and target gene expression than intraischemic hypothermia. However, with these cooling paradigms, we have previously shown that the extent of neuroprotection was similar (Han et al., 2002). This can be explained partially by the pathologic mechanisms involved in ischemic injury. NFκB is activated by reactive oxygen species (ROS), and ROS are especially increased during reperfusion. It is known that superoxide, a ROS, is decreased by hypothermia during reperfusion, but not intraischemically (Maier et al., 2002). Therefore, it is possible that cooling during this critical reperfusion period may decrease inflammatory gene transcriptional activity compared with intraischemic cooling. This suggests that the mechanism of protection may be different depending on the timing of cooling. The mechanism of protection by intraischemic hypothermia may be more related to preservation of metabolic stores, and suppression of glutamate release (early events) and delayed cooling during reperfusion may be more related to decreased ROS and a subsequent decrease in inflammatory gene expression at the transcriptional level.
Even if we consider IKK-related phosphorylation, subsequent ubiquitination, and degradation of IκB by the 26S proteasome as the main signal for NFκB activation, other regulatory systems may also be involved. As shown in Fig. 3D, the effect of hypothermia on IκB phosphorylation started at 6 h and became evident at 24 h after MCAO. However, we showed attenuated NFκB binding activity by hypothermia as early as 2 h after MCAO. This discrepancy in the time course between IKK action and NFκB binding suggests other mechanisms are involved in hypothermic suppression of NFκB activation. There are reports on NFκB translocation without degradation of IκB-α (Canty et al., 1999; Zwacka et al., 1998), and IKK is not the only enzyme involved in IκB turnover. Miyamoto et al. (1998) found that inhibitors of calpain, but not of the proteasome, ablated basal IκB turnover in WEHI 231 cells. Additional kinases have also been implicated in the regulation of IκB activity or stability. Protein kinase CK2 (previously known as casein kinase II) phosphorylates IκB on serines and threonines in the proline-glutamic acid-serinethreonine (PEST) sequence domain, which affects the intrinsic stability of this inhibitory protein (Barroga et al., 1995; Janosch et al., 1996; McElhinny et al., 1996; Schwarz et al., 1996; Shen et al., 2001). In addition, upstream mechanisms that control IKK can influence NFκB activation indirectly. To our knowledge, there have been no observations on these regulatory systems in ischemia models.
The evidence regarding whether NFκB potentiates or suppresses neural degeneration has been conflicting, since some target genes are known to be protective (e.g., manganese superoxide dismutase and various trophic factors) whereas others are damaging (e.g., iNOS, inflammatory cytokines). However, recent studies in stroke and cerebral ischemia models indicate that NFκB's actions are largely detrimental because mice deficient in the p50 subunit (Schneider et al., 1999) and animals treated with pharmacological NFκB inhibition (Phillips et al., 2000; Ueno et al., 2001) are both protected. However, the role of NFκB is far from definitive; one study showed that NFκB inhibition worsened ischemic injury (Hill et al., 2001). The data presented here showing suppression of NFκB by hypothermia is consistent with a damaging role.
In conclusion, we show that mild hypothermia has marked effects on gene expression at the transcriptional level through regulation of NFkB. Although activation of NFκB is an early step in the pathogenesis of cerebral ischemia, its long-lasting action makes it a good therapeutic target, particularly for postischemic, reperfusion-related ischemic injury. Mild hypothermia has been shown to have clinical efficacy in some settings (Bernard et al., 2002; Hypothermia After Cardiac Arrest Study Group, 2002); however, routine cooling of stroke victims may not always be practical or feasible. Future investigations should continue to focus on the mechanisms as well as the limits of protection in an effort to identify important pharmaceutical targets for treatment.
Footnotes
Acknowledgments:
The authors thank Dr. Daria Mochly-Rosen and Tamar Liron for their help with the IKK assays, Beth Hoyte for her assistance with the figures, and David Schaal for his help with manuscript preparation.