
Abstract
The authors examined the effects of pretreatment with 2-deoxy-D-glucose (2DG) on the middle cerebral artery occlusion–reperfusion (MCAO/R) model in hyperglycemic rats. Proton magnetic resonance imaging and spectroscopy were used to measure the lesion size, the level of cerebral perfusion deficit, and ratio of lactate to N-acetyl aspartate (NAA) in brain regions. By performing sequential diffusion weighted imaging, gradient echo bolus tracking, steady-state spin echo imaging, and water-suppressed proton magnetic resonance spectroscopy techniques, the time course of the early changes of the lactate/NAA peak ratio and perfusion deficit was examined in hyperglycemic rats undergoing 90-minute MCAO followed by 24-h reperfusion. Compared with the saline-treated hyperglycemic rats, 2DG treatment at 10 minutes before MCAO significantly reduced diffusion weighted imaging hyperintensity by ∼60% and the lactate/NAA peak ratio by ∼70% at 4 h after MCAO/R. Both spin echo–measured cerebral blood volume and dynamic gradient echo–relative cerebral blood flow showed that the restoration of blood supply recovered and remained at ∼80% of baseline during reperfusion in 2DG-treated hyperglycemic rats. These data suggest that inhibition of glucose metabolism by 2DG has a beneficial effect in reducing brain injury and minimizing the production of brain lactate during MCAO/R in hyperglycemic rats.
Keywords
The cerebral concentration and supply of glucose influence the outcome of cerebral ischemia. Elevated blood glucose at the onset of brain ischemia is well known to be associated with a worsened neurologic outcome (David and Bell, 1994; Huang et al., 1996; Li and Siesjo, 1997; Nedergaard and Diemer, 1987; Yip et al., 1991;). The detrimental effect of hyperglycemia on cerebral ischemia is considered to be mediated by the release of excitatory amino acids, additional formation of free radicals and enhanced accumulation of lactic acid, followed by a further decrease in intracellular and extracellular pH (Li and Siesjo, 1997; Li et al., 1999, 2000; Siesjo et al., 1990, 1993; Wei et al., 1997; Wei and Quast, 1998). By using 31P MRS, we observed that the preexisting hyperglycemic rat brain intracellular pH values were 6.14 ± 0.18 and 6.08 ± 0.19 during MCAO and reperfusion (Quast MJ, Wei J, Sell S, unpublished data, 1996), which is consistent with previous studies (Haraldseth et al., 1992; Suttle et al., 1992; Widmer et al., 1992) that showed hyperglycemia-related acidosis aggravates ischemic brain damage due to decreased intracellular and extracellular pH. It is assumed that lactate production in ischemic brain is a result of anaerobic metabolism of the higher concentration of available glucose (Li et al., 1994; Siesjo et al., 1993; Tyson et al., 1993). It has been reported that pretreatment with 2DG reduces brain damage and improves behavioral outcome in a normoglycemic rat focal ischemia model (Yu and Mattson, 1999) and reduces mortality and morbidity in hyperglycemic rats under the condition of four-vessel occlusion (Combs et al., 1986). 2-Deoxy-D-glucose exerted a cytoprotective effect preventing ischemia-induced delayed hippocampal neuronal damage in a gerbil transient forebrain ischemia model (Niwa et al., 1999). Enhanced epileptic tolerance in incomplete brain ischemia in mice (Rejdak et al., 2001) has also been reported. In addition, improved behavioral outcome and reduced degeneration of dopaminergic neurons in models of Parkinson disease has been noted (Duan and Mattson, 1999). In an in vitro study, 2DG suppression of synaptic transmission in the CA1 region of hippocampal slices was observed (Tekkok and Krnjevic, 1995). These data suggest that inhibiting energy-dependent processes can reduce hypermetabolic neuronal necrosis, which involves ATP depletion and lactate accumulation (Obrenovitch et al., 1988), glutamate excitotoxicity, and increased free radical formation. Here we test the hypothesis that by manipulating neuronal stores of glucose we can affect intracellular formation of lactate, thereby preventing the exacerbation of the brain lesion volume resulting from MCAO/R. The direct inhibition of glucose metabolism can be achieved by administration of the glucose analog 2DG, which competitively inhibits glucose uptake and entry into glycolysis. This glucose analog quickly enters the brain but is not metabolized beyond the hexokinase-catalyzed step and accumulates as a relatively stable 2DG6P. High levels of endogenous 2DG6P will cause inhibition of hexose phosphate isomerase (Horton et al., 1973). The goal of the present study was to assess the effect of pretreatment by 2DG on the pathophysiologic consequences of MCAO/R in hyperglycemic rats. Proton MRI and MRS were used to sequentially observe the size of the ischemic lesion and peak ratio of lactate/NAA after transient focal cerebral ischemia/reperfusion.
MATERIALS AND METHODS
Animal model
The University of Texas Medical Branch Animal Care and Use Committee approved all animal procedures. Male Sprague-Dawley rats (300 to 330 g body weight) were initially anesthetized with 3% isoflurane in balanced breathing air (30% O2 and 70% N2), and then intubated and mechanically ventilated with isoflurane maintained at 1.0% to 1.5% during the surgery and MRI/MRS procedures. The femoral artery was cannulated with a PE-50 polyethylene tube for monitoring blood gasses (PO2: 140 to 160 mm Hg, PCO2: 35 to 40 mm Hg, pH: 7.3–7.4) and mean arterial blood pressure. The cannula was inserted in the femoral vein to infuse glucose and deliver 2DG and MRI contrast agent. Rectal temperature was maintained at 37°C to 37.8°C with a water-circulated heating pad. Hyperglycemia was initially induced by a 1.0-mL bolus of 25% (% w/v) glucose through the femoral vein (1-minute duration) and continuous infusion at a rate of 60 μL/min (0.8 mmol/L per minute) maintaining a blood glucose level of 19 to 25 mmol/L (Patlak and Pettigrew, 1976). Glucose infusion was started 30 minutes before the MCAO and continued 1 h after reperfusion. Blood glucose concentration was measured during the period of baseline, MCAO, and 1 h into reperfusion. MCAO was induced using the intraluminal suture insertion method with some modifications (Huang et al., 1996; Longa et al., 1989; Nagasawa and Kogure, 1989). After an oblique incision on the right ventral lateral side of the neck, the ICA at the base of the common carotid artery bifurcation and the pterygopalatine artery (which is the only branch of the extracranial ICA) were ligated. An incision was made in the ICA, and a 1.5-cm length of 3–0 monofilament nylon suture coated with silicone was introduced into the lumen of the ICA and passed through the origin of the ipsilateral MCA, thereby blocking its blood supply. Reperfusion was allowed at 90 minutes after MCAO by the removal of the suture. The rats were divided into three groups: (1) hyperglycemic treated with 2DG (HR-2DG, n = 7); (2) hyperglycemic treated with saline (HR, n = 9); and (3) normoglycemic treated with saline (NR, n = 5). 2-Deoxy-D-glucose (300 mg/kg) was administered intravenously 10 minutes before MCAO. The animals were allowed to recover from anesthesia after the MRI/MRS procedure (∼4.5 h after reperfusion) and returned to their cages. They were reanesthetized for further study by MRI/MRS 24 h after MCAO/R. Individual experimental protocols varied in some cases, either because of technical problems or because hyperglycemic rats did not recover from the initial procedure.
Magnetic resonance imaging and spectroscopy experiments
Proton MRI was performed using a 4.7-T, 33-cm horizontal bore system (Varian, Palo Alto, CA, U.S.A.). Excitation and signal detection were achieved with a 5-cm surface coil. MRI and MRS were acquired before and 30 minutes after MCAO/R and repeated every 60 minutes up to 4.5 h and again at 24 h. The protocol consisted of several steps. First, water-suppressed, localized proton spectroscopy was obtained with a point-resolved spectroscopy sequence (PRESS), which was positioned in the ischemic territory. This 3.5 × 3.5 × 3.5 mm voxel contained caudate putamen and some overlying cortex (Fig. 1, top). The magnetic field over the voxel was homogenized using manual local shimming with the PRESS sequence, giving a line width at half height of the water signal of 10 to 15 Hz. The water signal was suppressed during observation with chemical shift-selective pulses optimized for detection of brain metabolites in the 1- to 4-ppm region. Three hundred acquisitions were obtained using a repetition time (TR) of 2,500 milliseconds and an echo time (TE) of 25 milliseconds. Multislice DWI, spin-echo T2WI, and PVI were acquired as 12 contiguous coronal slices covering a 20-mm length from the cerebellum to the olfactory lobe, with a 60 × 60-mm field of view, a 256 × 256 matrix, a TR of 3.0 seconds, a TE of 65 milliseconds, a slice thickness of 1.6 mm, and 128 phase encode steps. For the single-slice dynamic MRI in the area of the caudate putamen, a T2*-sensitive FLASH pulse sequence (Haase et al., 1986) with a 60 × 60-mm field of view was used. Each of the 40 frames was acquired with 64 phase encode steps, a TE of 3.0 milliseconds, a TR of 8 milliseconds (1.3 images/s), and one acquisition per phase encode step. A bolus of 0.3-mmol/kg gadopentetate dimeglumine (Magnevist) was injected, followed by 0.6-mL saline flushing into the femoral vein after the sixth frame. All animals were injected with Magnevist at the time points of baseline, 1 h into MCAO, and then 5 minutes, 1, 2, and 3 h after reperfusion. The PVI was only acquired at 4 and 24 h after MCAO/R, based on the T2-shortening effect of an SPIO nondiffusible intravascular tracer. The bolus of SPIO (2.0 mg Fe/kg body weight) was the same as that used for Magnevist injection.
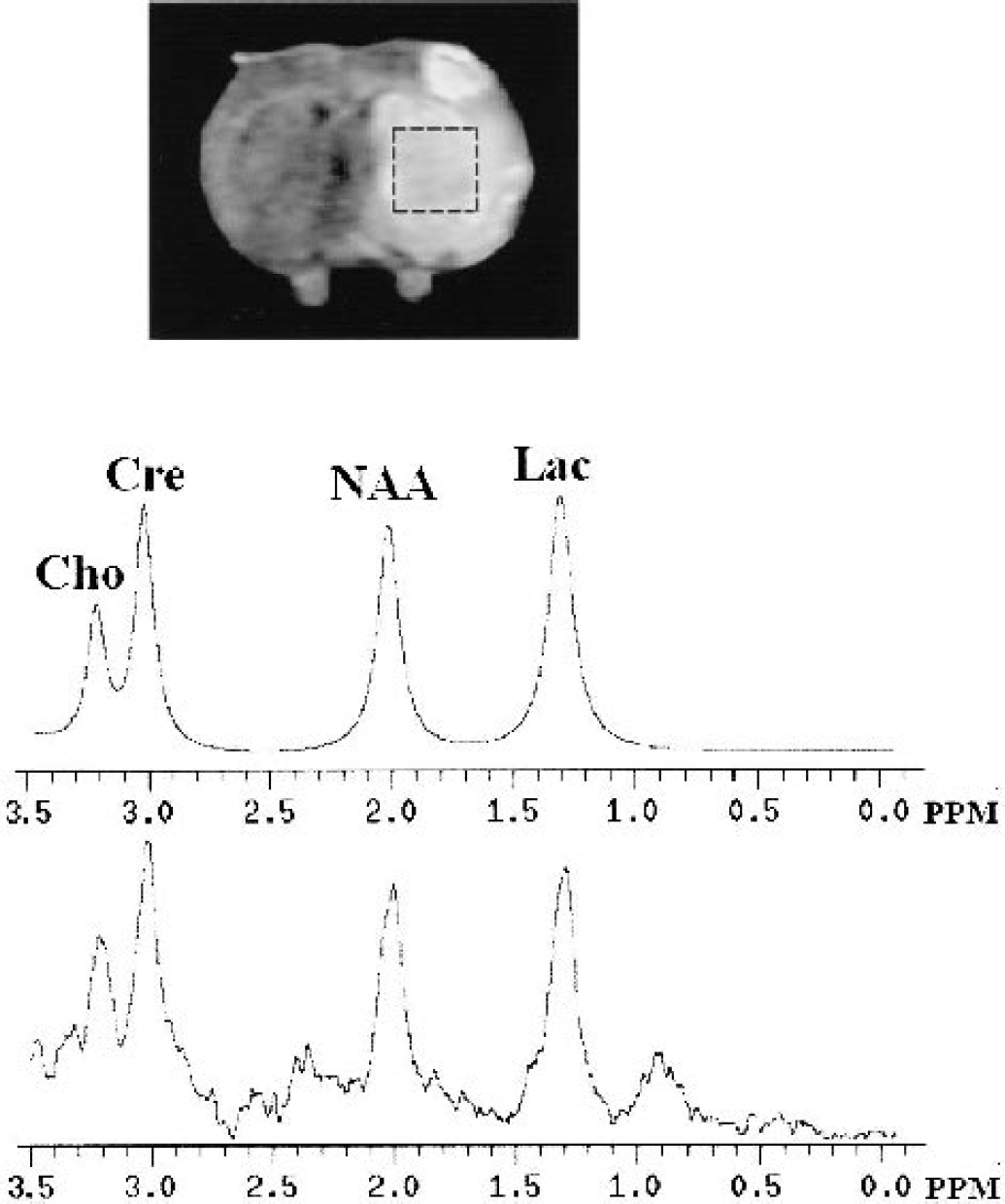
Representative in vivo 1H NMR spectrum (bottom) and fitted spectrum (middle) from one hyperglycemic rat. A point-resolved spectroscopy sequence in the dotted indicated region of cerebral infarction (top) with the voxel dimension (x, y, z) 3.5 × 3.5 × 3.5 mm3. The deconvolution spectrum fits the major brain metabolites: choline (Cho), creatine (Cre), N-acetyl aspartate (NAA), and lactate (Lac).
Data analysis
Ischemic lesions, estimated from DWI, were calculated based on intensity threshold segmentation using image analysis software developed in the Medical Imaging Laboratory at UTMB (Quast et al., 1993). Regions of restricted water diffusion measured in DWI were considered to represent cytotoxic edema (Knight et al., 1991; Moseley et al., 1990; Verheul et al., 1994), whereas regions of prolonged T2 relaxation time in T2WI represented vasogenic edema (Knight et al., 1991). The lesion area in each slice was multiplied by the slice thickness and summed to yield the total lesion volume in cubic millimeters. This technique using threshold values was used in previous studies in our laboratory (Huang et al., 1996; Quast et al., 1993, 1995, 1997; Wei et al., 1997; Wei and Quast, 1998). The region of the no-flow zone was estimated in a similar fashion using the signal-void region in PVI, which represented the area where no detectable intravascular tracer had perfused into the tissue (Kent et al., 1989, 1990; Quast et al., 1997). Single-slice digital image analysis was performed using indicator dilution methods (Axel, 1980; Quast et al., 1997; Zierler, 1962) to estimate CBF from CBV and mean transit time (MTT). Parametric images were constructed based on pixel-by-pixel calculation of hemodynamic parameters. Concentration–time curves representing the passage of tracer were constructed based on the observed signal intensity changes during the dynamic GE acquisition. Tracer concentration was determined from the following equation (Davis et al., 1989; Rosen et al., 1990):
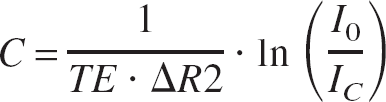
where C is the tissue concentration of Magnevist or SPIO iron tracer in the voxel or region of interest, ΔR2 is the proportionality constant relating signal intensity to tracer concentration at a given TE, IC is the signal intensity after tracer injection, and I0 is the baseline signal intensity. Gamma variate analysis was used to estimate curve parameters and correct for recirculation (Starmer and Clark, 1970; Thompson et al., 1964). The M1 of the tracer-concentration curves provides an estimate of the MTT (Weisskoff et al., 1993; Wittlich et al., 1995). Our regions of interest in the ipsilateral and contralateral forebrain are fed by the same main artery (the nonoccluded ICA feeding into the circle of Willis) and therefore have similar arterial input functions. Wittlich et al. (1995) showed the validity of this assumption using a similar MRI bolus tracking approach in rat MCAO models. Therefore, differences in M1 between ischemic and corresponding contralateral regions reflect differences in MTT. Relative MTT was estimated as follows:
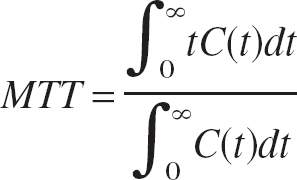
where C(t) is the time-dependent tracer concentration at time t after injection. Cerebral blood volume was estimated from the following equation (Wittlich et al., 1995):
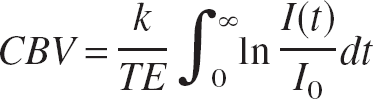
where k is a proportionality constant summarizing the arterial input function and ΔR2, I(t) is the time-dependent signal intensity, and I0 is the baseline signal intensity. Relative CBF was estimated as the ratio CBV/MTT.
The NMR spectral peaks were determined by manual integration using the standard Varian software in the MR system. The integral results were verified using spectral deconvolution. We assigned three peaks according to their positions referenced to the water signal at 4.76 ppm: the peak at 2.02 ppm was assigned to NAA, 1.33 ppm to lactate/lipid, and 3.02 ppm to creatine. Peak integrals of baseline-corrected proton spectra from the voxel of the ischemic lesion were used to measure the changes in lactate, creatine, and NAA. At baseline the peaks at 1.33 and 0.9 ppm were assumed to represent lipid species. During ischemia and reperfusion, the increase in area of the peak at 1.33 ppm was assumed to represent lactate. Using difference spectra to subtract out baseline peaks, we observed an increase only in the peak at 1.33 ppm. Lactate–lipid levels were expressed as the ratio of the lactate to NAA and as lactate to creatine.
Data are expressed as mean ± SD. Statistical differences were obtained using a repeated-measures analysis of variance, followed by a Bonferroni t-test for multiple group comparisons, using the statistical software SigmaStat (Jandel, San Rafael, CA, U.S.A.). Linear regression analysis was performed to determine the correlation between extent of DWI-measured brain injury and peak ratio of lactate to NAA. A value of P < 0.05 was considered to be significant for drug effects. Because of high mortality in the saline-treated hyperglycemic group, no statistical analysis was conducted on the 24-h data.
RESULTS
The physiologic variables for the three groups are shown in Table 1. There were no significant differences in these variables with the exception of blood glucose concentration, which was 19.6 ± 2.7 mmol/L in hyperglycemic rats compared with 4.5 ± 1.3 mmol/L in normoglycemic rats 10 minutes before MCAO. Hyperglycemic rats treated with 2DG showed a higher blood glucose concentration of 24.4 ± 4.2 mmol/L during the MCAO and 22.87 ± 3.7 mmol/L at 1-h reperfusion. There was 77.80% mortality by 24 h after ischemia in saline-treated hyperglycemic rats; 80% of 2DG-pretreated hyperglycemic rats and 87% of saline-treated normoglycemic rats survived more than 24 h after MCAO/R.
Major physiologic variables in the groups of rats studied
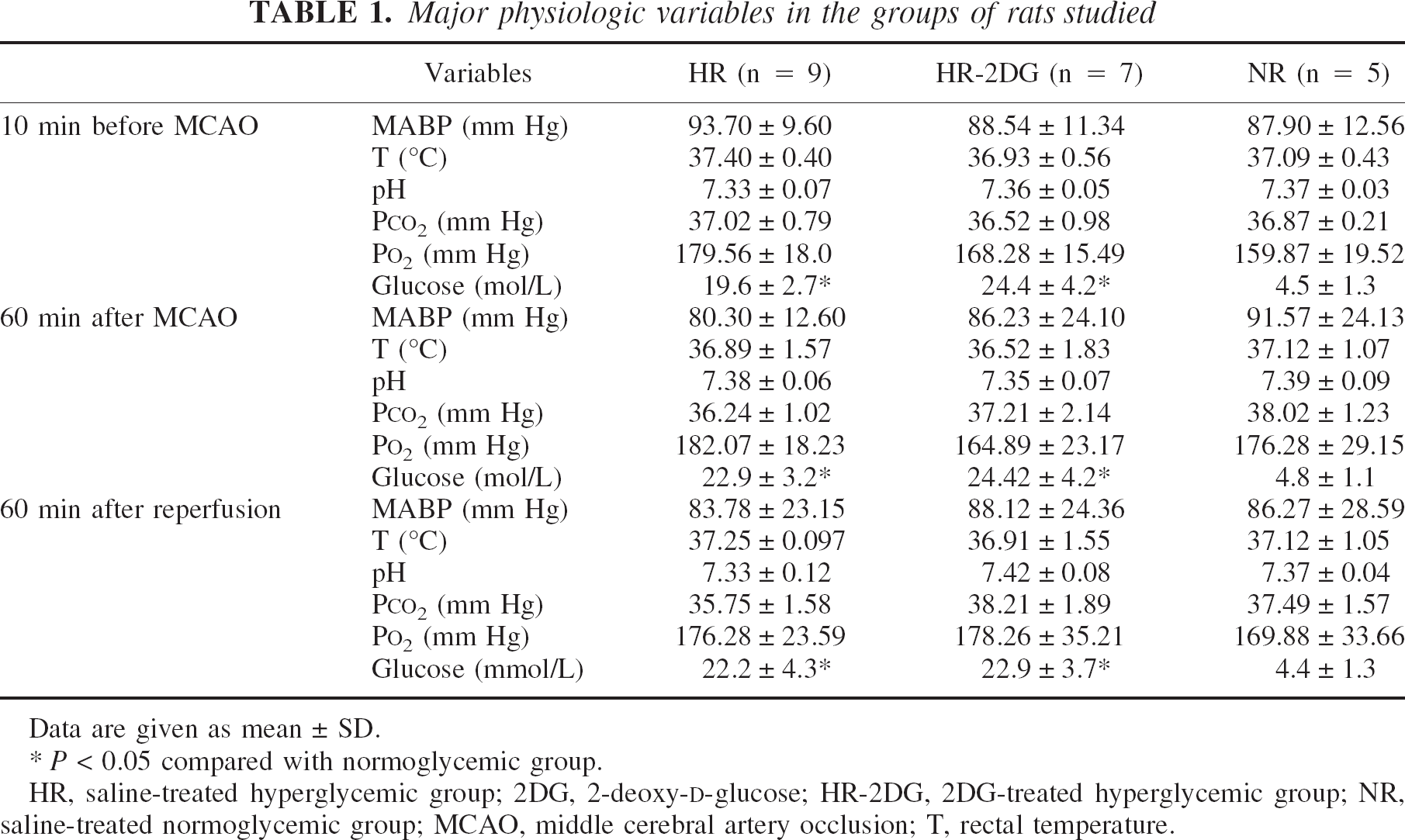
Data are given as mean ± SD.
P < 0.05 compared with normoglycemic group.
HR, saline-treated hyperglycemic group; 2DG, 2-deoxy-D-glucose; HR-2DG, 2DG-treated hyperglycemic group; NR, saline-treated normoglycemic group; MCAO, middle cerebral artery occlusion; T, rectal temperature.
The time course of changes in the lesion volume is summarized in Fig. 2. Pretreatment with 2DG exerted a significant neuroprotective effect by 1 h into reperfusion. In the NR and HR groups, the DWI and T2WI hyperintensity area was very similar to that reported previously (Quast et al., 1995, 1997). Development of cerebral lesions, observed as a high signal intensity area in the hyperglycemic rats (Fig. 3A), occurred not only in subcortical but also cortical brain regions. The hyperglycemic rats treated with 2DG showed the same patterns of lesion evolution and ischemic damage as the normoglycemic rats (Figs. 3B and 3C). Local reduction of brain damage by 2DG was prominent in ischemic lesions in the cortex. 2-Deoxy-D-glucose significantly reduced lesion volume (∼50%) in hyperglycemic rats compared with HR saline controls. By 4 h into reperfusion, DWI-detected lesion volumes were 140.95 ± 65.49, 202.31 ± 62.81, and 406.89 ± 112.17 mm3 for the NR, HR-2DG, and HR groups, respectively.
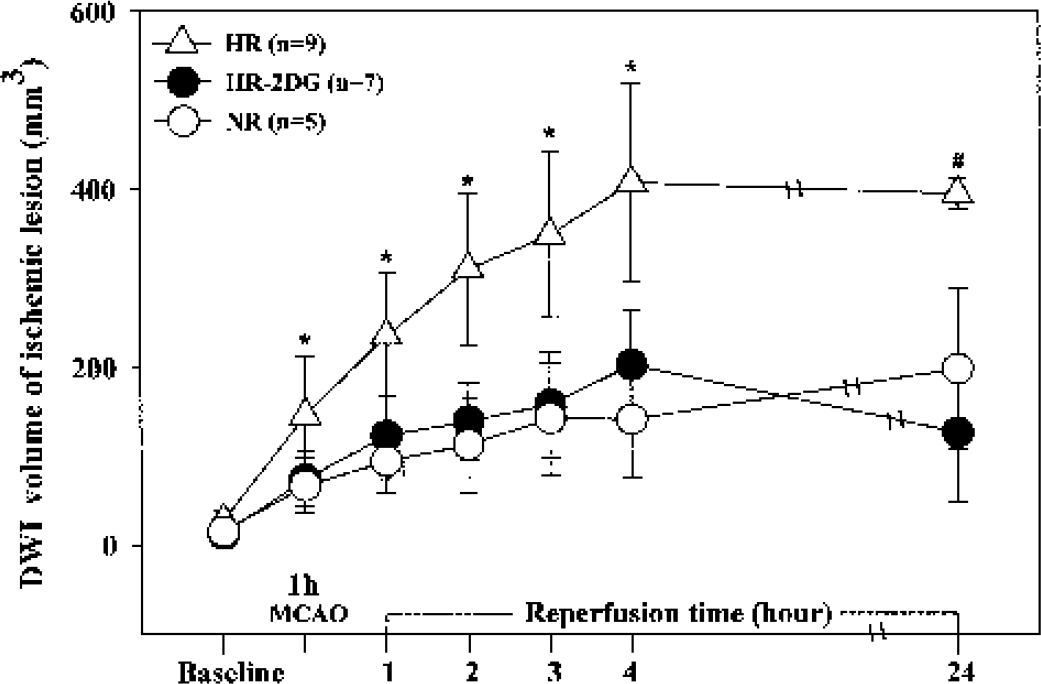
Time course of diffusion-weighted imaging–detected ischemic lesion volume (mean ± SD) in the total cerebral hemisphere. Triangle indicates hyperglycemic rats treated with saline (HR); black circle, hyperglycemic rats treated with 2-deoxy-D-glucose (HR-2DG); open circle, normoglycemic rats treated with saline (NR). *P < 0.05 compared with HR. #Data is for two of nine rats owing to high mortality in the HR group. MCAO, middle cerebral artery occlusion.
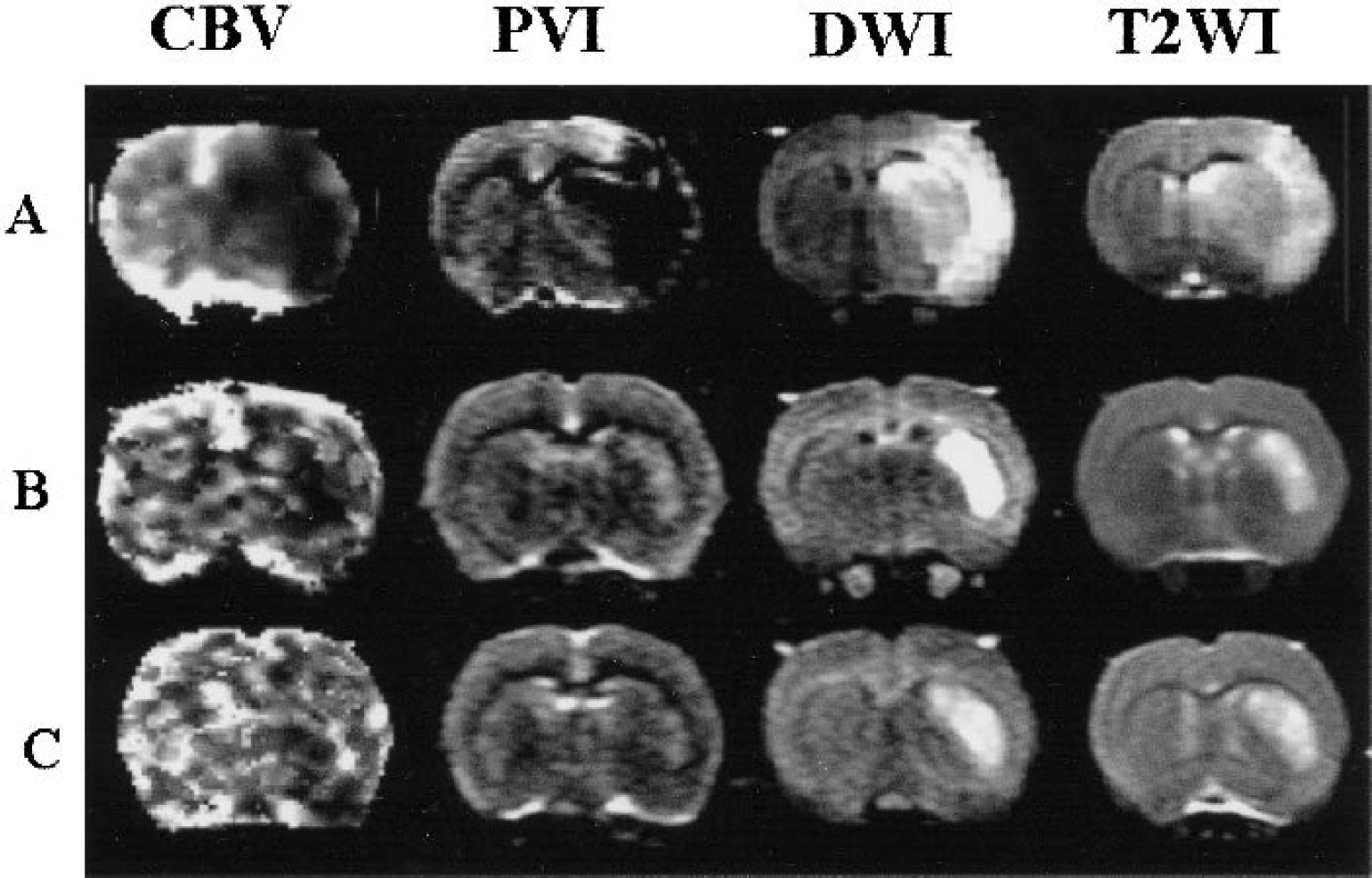
Representative cerebral blood volume, plasma volume imaging, diffusion-weighted imaging, and T2-weighted imaging images at the level of the caudate putamen. Images were acquired at 5.5 h into middle cerebral artery occlusion–reperfusion.
The MRI-measured relative CBF in the ischemic and control hemispheres is summarized in Fig. 4. There was no significant difference in the residual CBF (∼50% of normal baseline) among the three groups at 1 h into MCAO. Although the restoration of blood supply produced immediate tissue reperfusion in all experimental groups, the level of CBF recovery was limited and temporary in the HR saline group and remained at ∼57% to 60% of the intact-side rates during perfusion. Half of the rats in both the NR and HR-2DG groups showed minor declines in perfusion over time in CBF and the others showed hyperperfusion. Figure 5 shows the relative PVI acquired 4 and 24 h after MCAO/R from a representative rat in each of three experimental groups. In PVI or CBV images, signal intensity is proportional to the volume of circulating blood, so areas with a perfusion deficit are represented by low signal intensity. Hyperglycemic rats treated with saline showed very low intensity in the right hemisphere by PVI, indicating the great extent of injury. In contrast, a normal- or high-intensity area of PVI was detected in the right hemisphere in both of the NR and HR-2DG groups, thus confirming improved tissue reperfusion throughout the previously ischemic hemisphere. Typical DWI, T2WI, PVI, and perfusion images of one rat from each experimental group at the level of the caudate putamen at 5 h into MCAO/R are shown in Fig. 3. Brain tissue injury appears as hyperintensity in DWI and T2WI but low signal intensity in PVI and CBV images. Comparison of the three groups by MRI indicated that 2DG reduced ischemic injury in the hyperglycemic group.
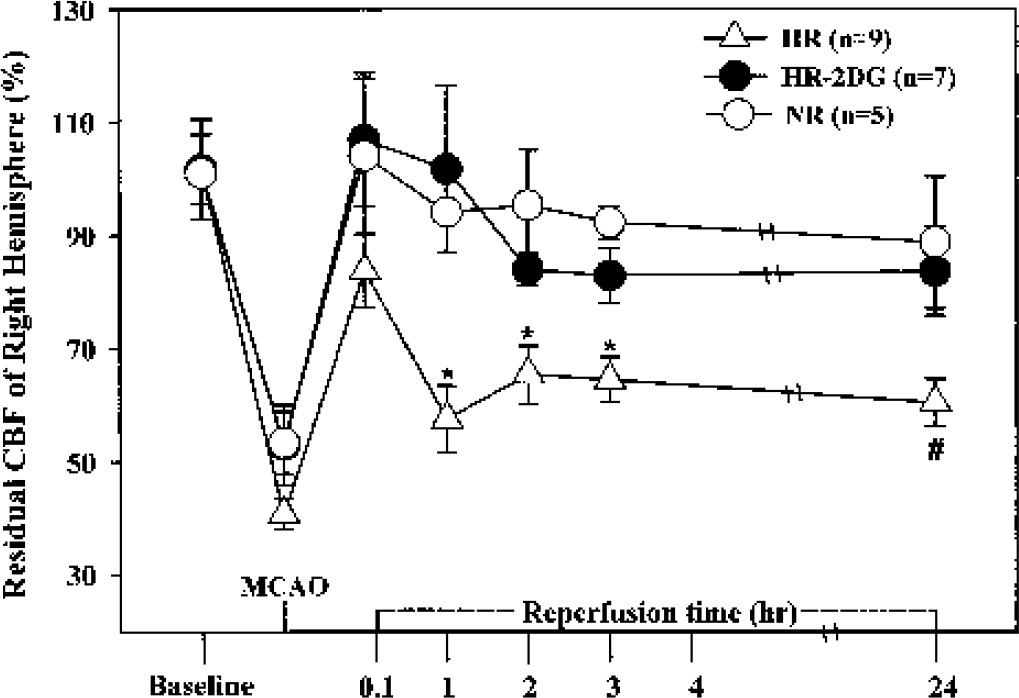
The time course of percent changes in residual cerebral blood flow of the right hemisphere. Triangle indicates hyperglycemic rats treated with saline (HR); black circle, hyperglycemic rats treated with 2-deoxy-D-glucose (HR-2DG); open circle, normoglycemic rats treated with saline (NR). *P < 0.05 compared with HR. #Data is for two of nine rats owing to high mortality in the HR group. MCAO, middle cerebral artery occlusion.
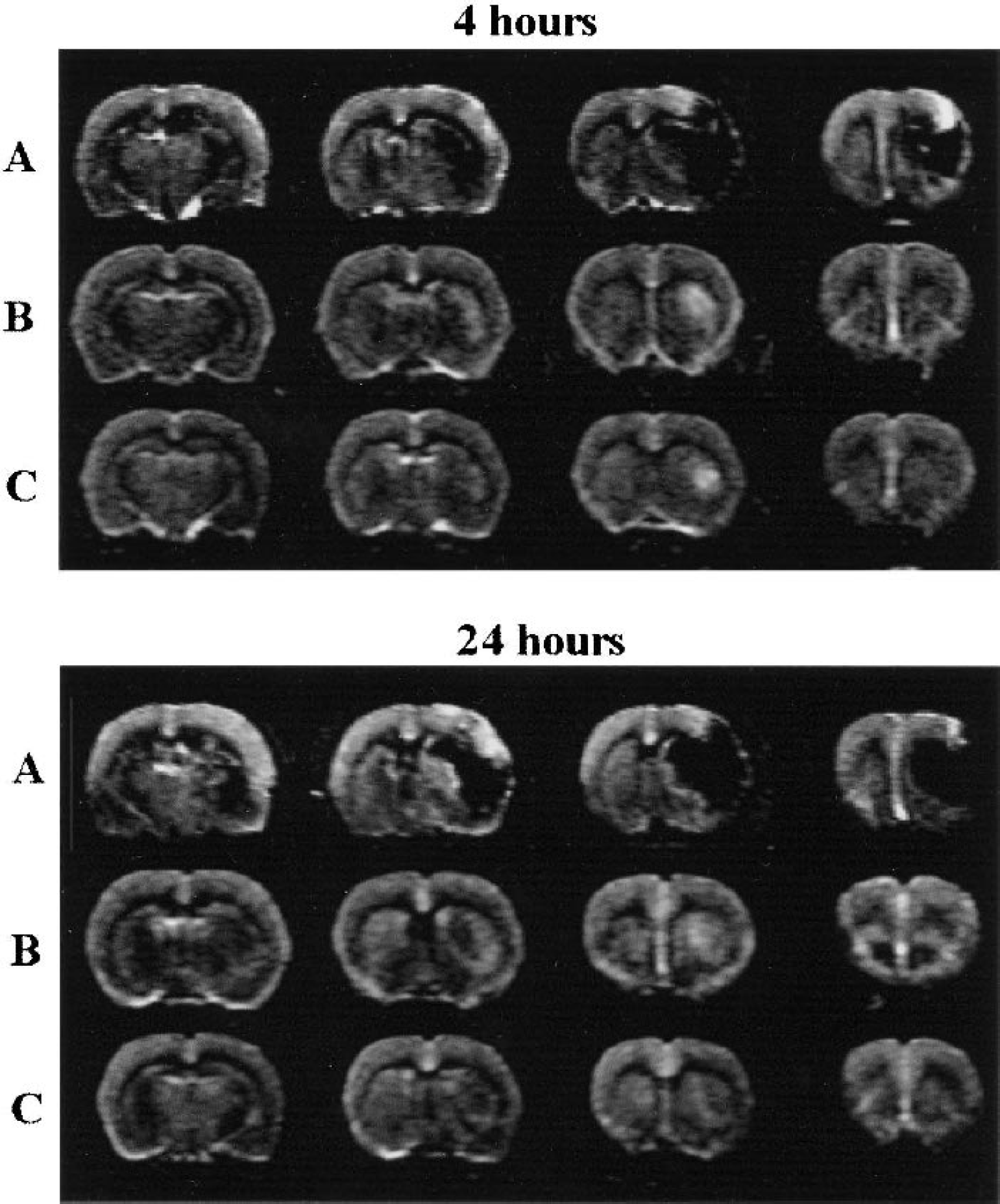
Consecutive coronal forebrain relative plasma volume images at 4 and 24 h after middle cerebral artery occlusion–reperfusion (MCAO/R). Saline-treated hyperglycemic rats had a significant no-reflow zone (low intensity) in the MCA territory
There were no significant differences in 1H spectra among the three groups during the period of baseline and MCAO. Intrasubject variability of the NAA peak showed a standard deviation of 18.3% from baseline through 4 h after reperfusion. After 1 h of reperfusion, 1H spectra showed a significant increase in lactate in HR rats with a moderate (nonsignificant) decrease in NAA. After 2 h, all of the experimental groups MRS in the central ischemic region showed a marked increase in lactate and peak ratio of lactate to NAA. An elevated lactate to NAA ratio was shown to be associated with a larger area of MRI-detected cerebral lesion as well as a larger perfusion deficit. The lactate to NAA ratio was significantly lower in the 2DG-treated HR group compared with the HR group by 2 h into reperfusion. Peak ratios of lactate to NAA and lactate to creatine are shown in Table 2. A summary of linear regression analysis with ρ = 0.95 indicates a good agreement between elevated peak ratio of lactate to NAA and extent of DWI-measured brain injury in the three experimental groups.
Ratio of lactate to N-acetyl aspartate and lactate to creatine for three experimental groups
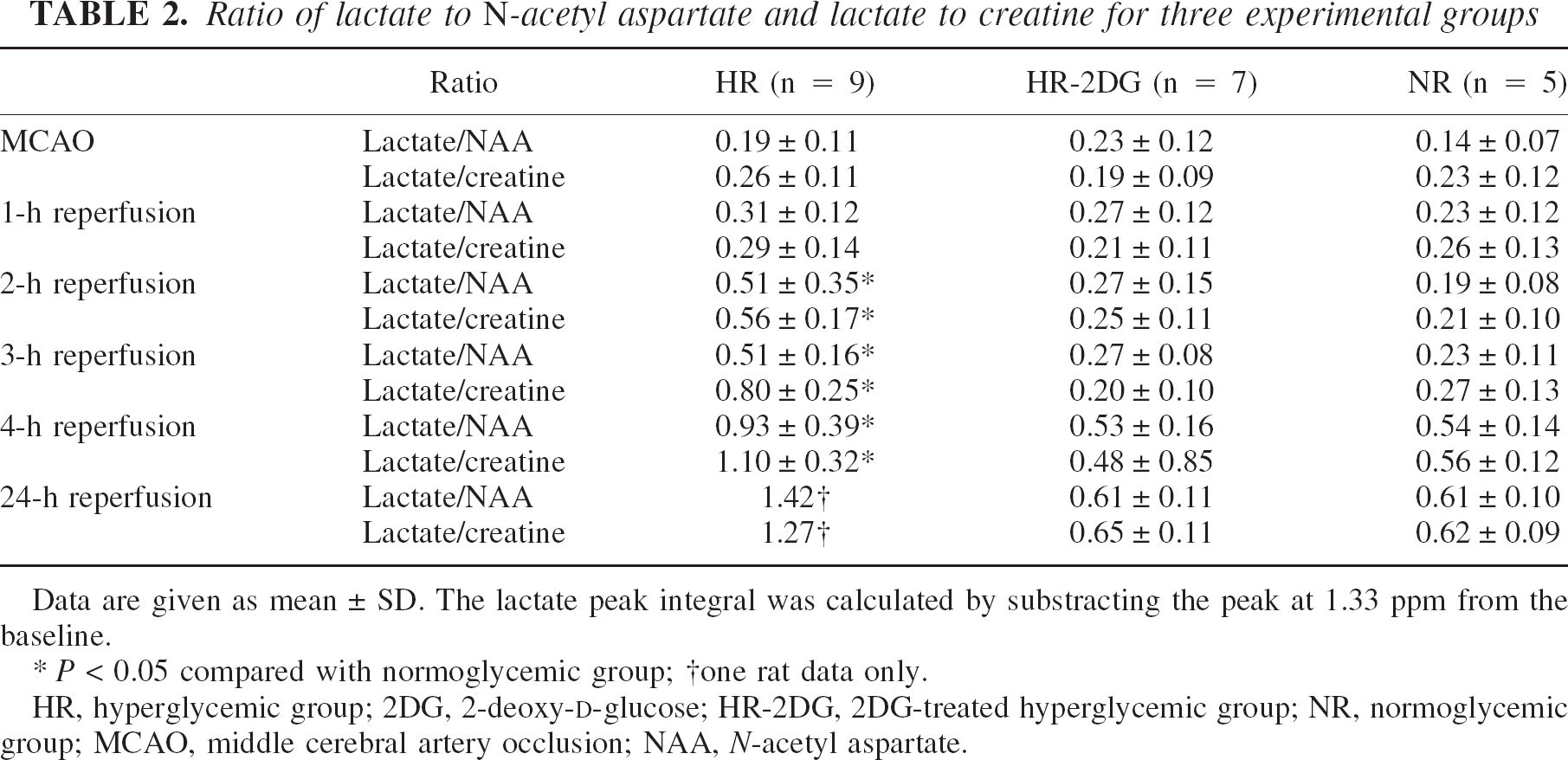
Data are given as mean ± SD. The lactate peak integral was calculated by substracting the peak at 1.33 ppm from the baseline.
P < 0.05 compared with normoglycemic group
one rat data only.
HR, hyperglycemic group; 2DG, 2-deoxy-D-glucose; HR-2DG, 2DG-treated hyperglycemic group; NR, normoglycemic group; MCAO, middle cerebral artery occlusion; NAA, N-acetyl aspartate.
DISCUSSION
The present study shows that pretreatment with 2DG is neuroprotective in the hyperglycemic rat model of suture-inserted MCAO/R. 2-Deoxy-D-glucose–pretreated hyperglycemic rats showed reduced lactate levels accompanied by beneficial effects minimizing the neuropathologic consequences of ischemic injury. By using in vivo 1H NMR and MRI, the present results support the hypothesis that a large cerebral infarct is associated with raised lactate and reduced NAA concentration as well as critical perfusion deficit. Lactate concentrations are normally low in the brain (near the MRS detection limit in vivo) and, as the end product of anaerobic glycolysis, are a particularly useful measure of challenged metabolism. The degree and extent of tissue damage is correlated with the content of lactic acid (Graham et al., 1993; Widmer et al., 1992), and an increase in morbidity and mortality in hyperglycemia has been linked to excessive lactate production and the resulting acidosis (Pulsinelli et al., 1982; Siemkowicz and Gjedde, 1980). Our previous reports (Huang et al., 1996; Quast et al., 1995, 1997; Wei et al., 1997; Wei and Quast, 1998) are consistent with this concept. In the HR and NR groups, blood glucose levels correlated with the size of lesion volume 5 h after MCAO/R (ρ = 0.902) and with intracerebral lactate levels measured in vivo (ρ = 0.879). In 2DG-treated rats, lactate levels correlated with infarct volume but not with blood glucose levels. Rothman et al. (1990) used 1H MRS- and 13C-labeling methods to show that persistently elevated lactate levels in a region of cerebral infarction were derived from active anaerobic metabolism of glucose. All of these findings are consistent with an earlier suggestion by Siemkowicz and Gjedde (1980) that the damaging factor in the hyperglycemic group is increased lactic acidosis associated with prolonged anaerobic glycolysis. Because all the cerebral lactate arises from glycolysis of serum glucose, hyperglycemia-exacerbated cerebral acidosis during MCAO/R and subsequent lactate accumulation are dependent on glucose concentration (Widmer et al., 1992). Yu and Mattson (1999) also reported that daily treatment with 2DG in normoglycemic rats reduced focal ischemic brain damage due to a decreased level of glucose available to neurons over time. A subtoxic dose of 2DG inhibits cellular metabolism by competitively inhibiting glucose uptake and anaerobic glycolytic flux, and may exert cytoprotection of brain tissue against otherwise lethal insults.
A rise in the extracellular concentration of EAAs plays a pivotal role in hyperglycemic rats under ischemic conditions (Wei and Quast, 1998). Gomi et al. (2000) reported that, with MCAO, reduced EAA uptake was observed with elevated local cerebral glucose metabolism in the region as well as local cerebral blood flow of 30% to 50% of the intact side in a rat study. We also observed that hyperglycemic rats treated with saline showed temporary blood flow recovery during reperfusion, followed by a decline to a level 57% to 60% of baseline over time. Under this condition, persistent anaerobic glycolysis occurred resulting in lactate production. Anaerobic glycolysis may persist after transient ischemia in spite of the recovery of local cerebral blood flow to a level that is greater than the threshold for the activation of anaerobic glycolysis (less than 40% of the control) (Tanaka et al., 1985). Our previous study (Wei and Quast, 1998) reported that exacerbation of postischemic brain injury observed in hyperglycemic rats is associated with enhancement of EAA release (or reduction of EAA reuptake), toxic metabolism of NO, and hydroxyl radical formation under acidosis conditions. Free radical formation induced by brain ischemia is involved in the development of infarction and vasogenic edema. During ischemia, brain production of free radicals causes an increased output of EAA. Our previous studies (Wei et al., 1997; Wei and Quast, 1998) in the hyperglycemic rat showed that, after MCAO/R, hydroxyl free radical concentration increases steadily with time up to two- to threefold of baseline. Volterra et al. (1994) provided evidence for an almost linear decline of glutamate transport during exposure of cortical astrocytic culture to free radicals, suggesting that the EAA transporters are important targets of reactive oxygen species. Pellegrini-Giampietro et al. (1990) observed a significant release of EAA after incubation of hippocampal slices with enzymes and substrates known to cause the formation of free radicals.
It has been hypothesized that there is a pathologically vicious circle among EAA, NO, and free radicals due to ischemia-induced acidosis (Gomi et al., 2000; Pellegrini-Giampeitro et al., 1988, 1990; Wei and Quast, 1998). NO-induced cell death is probably triggered by inhibition of cellular respiration, followed by glutamate release and excitotoxicity. In vitro evidence (Bal-Price and Brown, 2001) indicated that NO donors caused rapid ATP depletion in neuronal cultures followed by activation of glycolysis with increased lactate levels and acidification of the culture medium. Decreased levels of L-lactic acid (and increased pH) in the presence of NO synthase inhibitor suggested that NO mediated the activation of glycolysis. Taken together with our previous results (Huang et al., 1996; Quast et al., 1995, 1997; Wei et al., 1997; Wei and Quast, 1998) and demonstration of a similar degree of neuroprotection by inhibition of NO synthesis, we suspect there may be a link between excess NO-induced EAA excitotoxicity and glucose metabolism.
Numerous studies have focused on the association between high lactate concentration in the brain and cerebral injury. Earlier studies (Petito et al., 1987) reported that injection of sodium lactate solution with pH 4.5 produced neuronal necrosis as soon as 1 h after injection, followed by necrosis of astrocytes and intravascular thrombi at 3 and 6 h. Injectants produced brain necrosis in a histologic pattern resembling ischemic infarction only when pH was less than or equal to 5.30 (Kraig et al., 1987). Thus, rather than lactate itself being the toxic intermediate, it seems more likely that the acidic environment is the more detrimental condition. This is corroborated by previous results in the hyperglycemic MCAO/R model where 31P MRS measured an intracellular pH of 6.10 compared with 6.90 in normoglycemic counterparts. Nonetheless, while probably not a major neurotoxic agent in itself, accumulated lactate does seem to be a strong indicator of hypermetabolic necrosis and correlates with the severity of irreversible brain damage.
Agents such as 2DG inhibit hypermetabolic neuronal necrosis (which involves lactate accumulation, glutamate excitotoxicity, NO production, free radical formation, and ATP depletion) and limit irreversible brain damage by suppressing the energy-dependent pathologically vicious cycle. The present investigation is, to our knowledge, the first to evaluate a nonmetabolizable glucose analog (2DG) on the hyperglycemic rat MCAO/R model. This study clearly shows that 2DG is effective in minimizing hyperglycemic exacerbation of ischemia–reperfusion brain injury and in tempering the rise in brain lactate, as evidenced by an MRS-detected decrease of the lactate to NAA ratio in the affected region. These results provide evidence that pretreatment with 2DG can reduce brain injury and lactate production.
Footnotes
Acknowledgments:
The authors thank Jose M. Gonzalez for his assistance with MRI techniques, and Edward L. Ezell for his assistance with manuscript preparation.