
Abstract
Cerebrovascular abnormalities, in endothelium and smooth muscle compartments, occur in the course of cerebral ischemia–reperfusion as evidenced by the impairment of endothelium-dependent relaxation and decrease in potassium inward rectifier density in occluded middle cerebral arteries (MCAs). The authors investigated whether a delayed vascular protection occurred in a model of brain ischemic tolerance. A low dose of lipopolysaccharide (0.3 mg/kg) administered 72 h before MCA occlusion induced a significant decrease in infarct volume. In parallel to this delayed neuroprotective effect, lipopolysaccharide prevented the ischemia–reperfusion–induced impairment of endothelium relaxation. In addition, lipopolysaccharide prevented the postischemic alteration of potassium inward rectifier–dependent smooth muscle relaxation as well as the decrease in potassium inward rectifier density measured by patch-clamp in dissociated vascular smooth muscle cells originated from the occluded MCA. These results suggest that during brain ischemic tolerance, lipopolysaccharide is able to induce both a delayed neuroprotective and vasculoprotective effect.
Keywords
Several findings suggest that cerebrovascular abnormalities can occur in the course of cerebral ischemia–reperfusion. Loss of vascular tone, diminished responsiveness to the vasoconstrictor effect of 5-hydroxytryptamine (5-HT), abolition of the endothelium-dependent relaxation in response to acetylcholine, and diminished distensibility have been reported (Cipolla et al., 1997; Rosenblum et al., 2000). Deleterious effects of ischemia–reperfusion also occur in the smooth muscle of the occluded cerebral artery where alterations of barium-sensitive K+ inward rectifier (KIR) channel currents, which are involved in the relaxation of cerebral arteries, have been described (Bastide et al., 1999; Marelli et al., 1998). By their consequences for cerebral blood flow, these vascular abnormalities could affect, in turn, the neuronal function. Indeed, we have previously demonstrated a significant correlation between ischemia-induced decrease of KIR channel density in the occluded artery and the severity of brain injury and brain edema (Bastide et al., 1999). These data suggest that vascular protection could also be a new pharmacological approach in parallel to neuroprotection.
Oxidative stress has been suggested to play an essential role in vascular injury after ischemia–reperfusion. The vascular alterations are related to the reperfusion rather than to the ischemia since cerebral artery occlusion alone fails to induce such changes (Cipolla et al., 1997). Reperfusion-induced reactive oxygen species (ROS) contribute to a decrease of the nitric oxide (NO) availability responsible for the postischemic endothelial dysfunction (Drexler, 1999). In addition, ROSs have a direct deleterious effect against ionic channels (for review, see Kourie, 1998). This role of oxidative stress suggests that its pharmacological modulation could contribute to prevent postischemic cerebrovascular abnormalities. It is now well known that there is a possibility of protecting the brain against ischemia–reperfusion by different procedures like ischemic preconditioning (Chen and Simon, 1997) or lipopolysaccharide (LPS) administration (Bordet et al., 2000; Tasaki et al., 1997). The procedures, such as preconditioning (Chen and Simon, 1997) or LPS administration (Dawson et al., 1999), inducing a delayed neuroprotection (so-called brain ischemic tolerance) by interaction with brain oxidative status and NO pathway could contribute to prevent post-ischemic cerebrovascular impairment. But the question remains controversial about the involvement of changes of regional cerebral blood flow (Dawson et al., 1999; Matsushima and Hakim, 1995).
We have previously shown that a low dose of LPS induced a delayed neuroprotection in parallel to a delayed increase in endogenous brain superoxide dismutase activity (Bordet et al., 2000). Moreover, the same low dose of LPS induces a delayed enhanced NO-mediated relaxation in rat peripheral arteries (Pu et al., 1999) and an increase of endothelial NO synthase expression in the endothelium of cerebral blood vessels (Puisieux et al., 2000), both of which could underlie a vascular protective effect in course of cerebral ischemia–reperfusion. We have so hypothesized that a low dose of LPS could induce a delayed vascular protection in parallel to its delayed neuroprotective effect. Thus, the aim of this study was to evaluate the preventive protective effect of a low dose of LPS administered 72 h before middle cerebral artery (MCA) occlusion, against both postischemic endothelial dysfunction and KIR alteration.
MATERIALS AND METHODS
All experiments were performed in strict accordance with the guidelines of the National Institutes of Health and French Department of Agriculture.
Animals and drug administration
Lipopolysaccharide (0.3 mg/kg) derived from Escherichia coli (serotype 055: B5; 1 mg LPS represents 2.6.106 UI endotoxin) or vehicle (saline 0.9%) were administered intraperitoneally to male Wistar rats (IFFA Credo, L'Arbresle, France), weighing 280 to 320 g, 72 h before MCA occlusion or sham surgery. Four groups of rats were defined: (1) vehicle-treated sham (SHAM + VEH): (2) vehicle-treated ischemic–reperfused (I/R + VEH); (3) LPS-treated sham (SHAM + LPS); and (4) LPS-treated I/R (I/R + LPS).
Middle cerebral artery occlusion
Anesthesia was induced by chloral hydrate administered intraperitoneally at a dose of 300 mg/kg. A rectal probe was inserted and body temperature was maintained with a heating lamp at 37°C ± 0.5°C. The caudal artery was exposed and cannulated with a 24-G polyethylene catheter and connected to a blood pressure monitor. Mean arterial blood pressure (mm Hg) was monitored throughout the experiment and blood samples were taken before, during, and after ischemia to measure blood pH and arterial Pa
The ostium of the right MCA was occluded intraluminally as previously described (Bastide et al., 1999). The right carotid arteries were exposed through a midline cervical incision and the common carotid and external carotid arteries were ligated with a silk suture. The pterygopalatine artery was exposed by developing a plane alongside the internal carotid artery, and was ligated at its origin with a fine silk. Aneurysm clip was placed across internal carotid artery and an arteriotomy was made in the common carotid artery stump allowing the introduction of a 4/0 monofilament nylon suture with a tip that was rounded by flame heating. This was secured in place and the aneurysm clip on the internal carotid artery was removed. The suture was gently advanced into the internal carotid artery and passed into the intracranial circulation to lodge in the narrower lumen of the origin of the MCA. Mild resistance to this advancement indicated that the intraluminal occluder had entered the anterior cerebral artery. After 60 minutes, the suture was carefully removed, until its tip was blocked by ligature placed on common carotid artery, to permit reperfusion. The caudal artery catheter was removed and the artery was ligated to prevent bleeding. The animals were placed in cage to recover from anesthesia at room temperature and were allowed to eat and drink freely.
Histology and measurement of infarct size
Twenty-four hours after reperfusion, the rats were killed with an intraperitoneal overdose of pentobarbital (200 mg/kg) and brains were rapidly removed in an ice-cold Krebs solution (in mmol/L: NaCl 119; NaHCO3 24; KCl 4.7; KH2PO4 118; MgSO47H2O 1.17; glucose 10; CaCl2 1.6) gassed with 5% CO2 and 95% O2. Middle cerebral arteries were carefully dissected to the vascular smooth muscle cell (VSMC) preparation or for vasoreactivity analysis; thereafter, brains were frozen and coronally dissected into 50 μm-thick slices on a cryostat at 12 levels separated by 1-mm intervals, according to stereotaxic sections maps (Paxinos and Watson, 1986). Sections were stained with cresyl fast violet, and the unstained area of the brain was defined as infarcted. Cortical and subcortical infarcted areas and total hemispheric areas were calculated separately for each coronal slice with an image-analysis software (Color Image 1.32, NIMH, Bethesda, MD, U.S.A.) after digitization by a scanner process. Total infarct and hemispheric volumes (in mm3) were calculated by the use of numerical integration of the respective areas for all the sections per animals and the distance between them. A corrected total infarct volume was calculated to compensate for the effect of brain edema (Lin et al., 1993). The corrected volume was calculated using the following equation: Corrected infarct volume = Total infarct volume – (Right hemisphere volume – Left hemisphere volume). The edema volume was represented by the difference between right and left hemisphere volumes.
Electrophysiologic study
All experiments were done on freshly dissociated cerebral VSMC obtained from MCAs ipsilateral to occlusion in ischemic rats and from MCAs of sham animals. Middle cerebral artery myocytes were obtained by an enzymatic procedure described elsewhere (Bastide et al., 1999). Cells were placed in Petri dishes mounted on the stage of a phase contrast inverted microscope. The whole-cell patch-clamp technique (Hamill et al., 1981) was used to evaluate KIR current by using a RK 400 patch-clamp amplifier (Bio-Logic, Claix, France) connected to a microcomputer equipped with pClamp 5.5 software (Axon Instruments, Foster City, CA, U.S.A.). The currents were low-pass filtered at 3 kHz, digitized at 10 kHz using a 12-bit analog-to-digital converter (Labmaster, TL-1, Scientific Solutions, Solon, OH, U.S.A.). The membrane capacitance of the cells was measured by integrating the area of capacitive transient elicited by a 5-mV depolarizing pulse from a holding potential of −50 mV. The pipette capacitance was not subtracted from total membrane capacitance and there was no leak substraction. The pipettes were pulled from thin borosilicate glass capillaries (Clark Electromedical Instruments, Reading, U.K.) and had a resistance of 2.7 to 3.3 MΩ (when filled with the pipette solution containing in mmol/L: 130 KCl, 2 MgCl2, 10 EGTA, 10 HEPES, and 5 phosphocreatine; pH 7.3). The bath solution contained (in mmol/L): 134 NaCl, 6 KCl, 1 MgCl2, 0.1 CaCl2, and 10 HEPES (pH 7.4). Control and test solutions were applied to the exterior of the cell by placing the cell at the opening of 300-μm-inner-diameter catheters fixed on the motorized rotating head of a RSC 200 (Rapid Solution Changer; Bio-Logic). All experiments were done at room temperature (19°C to 22°C).
The standard voltage clamp protocol consisted of 185 ms-voltage ramps from −140 to 50 mV, from a holding potential of −60 mV. In each experiment, currents in response to four voltage ramps were averaged to give the final values used in this work. The current density was calculated and preferentially used to compare among the different cells to eliminate variation of the cell size.
Vasomotricity analysis
Segment of dissected right MCA was mounted in a small vessel arteriograph (Living Systems Instrumentation, Burlington, VT, U.S.A.) on two glass cannulas perfused with Krebs solution. The artery was secured on the proximal and distal cannulas with nylon ties. The distal cannula was closed in order to work in a no-flow condition. The arteriograph chamber was continuously supplied with Krebs solution equilibrated with 5% CO2/95% O2 and maintained at 37°C and pH 7.4. The proximal cannula was connected to a pressure transducer, a miniature peristaltic pump, and a servo controller that continually measured and adjusted transmural pressure to 20 mm Hg for 15 minutes. The entire arteriograph system was positioned on the stage of an inverted microscope equipped with a video camera and a monitor. The lumen diameter was measured by image analysis with a video dimension analyzer. The perfusion pressure was slowly increased to 60 mm Hg, and the MCAs were allowed at least 1 h to stabilize before experiments were conducted.
To test the endothelium-dependent and smooth muscle-dependent relaxations, two protocols were successively applied. First, serotonin (5-HT) at a concentration of 10–6 mol/L, which induced 90% of the maximum constriction, was applied to the vessel in the Krebs solution. When diameter reached a steady state, acetylcholine (10–5 mol/L) or sodium nitroprus-side (SNP 3.10–5 mol/L) was added to the 5-HT–containing Krebs solution until a steady dilation was reached. Vasorelaxation is expressed as the percentage of increase of the preconstricted artery diameter. Second, after washout with fresh Krebs solution and the return to passive diameter, a 15-mmol/L KCl-containing Krebs solution was superfused to the vessel to achieve a KIR smooth muscle–dependent vasodilation, which is inhibited by administration of BaCl2 (75 μmol/L), a specific KIR 2.1 blocker. Vasorelaxation was estimated as the percent increase of the passive diameter.
Statistical analysis
All data are expressed as mean ± SD. Continuous variables (infarct volumes, superoxide dismutase [SOD] dosage, KIR densities, MCA relaxations) were compared with a one-way ANOVA and, when significant, followed by a post hoc PLSD Fisher test. A value of P < 0.05 was considered significant.
RESULTS
Lipopolysaccharide effects on infarct size
Pretreatment with LPS (0.3 mg/kg), administered intraperitoneally 72 h before MCA occlusion, resulted in a significant decrease of 34% in the infarct volume (131 ± 54 mm3; n = 12) when compared with the infarct volume of the vehicle-treated group (199 ± 69 mm3; n = 10; P < 0.01) (Fig. 1).
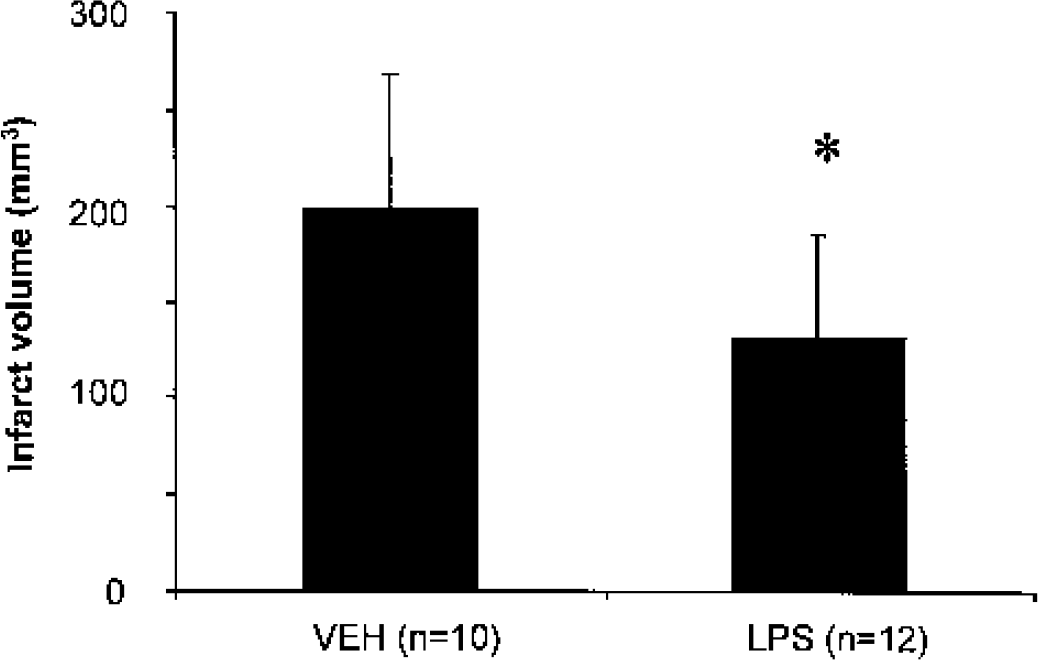
Effect of lipopolysaccharide (LPS) on total infarct volume (corrected for edema) induced by 1-h focal cerebral ischemia. Values are mean ± SD; *P < 0.05 compared with vehicle (VEH).
Lipopolysaccharide effects on endothelium-dependent vasoreactivity
There was no difference in 5-HT–induced contractile response among the four groups (Table 1). In MCAs from SHAM + VEH and SHAM + LPS animals, acetylcholine (10–5 mol/L) induced a similar endothelium-dependent relaxation after a 5-HT preconstriction (10–6 mol/L). In I/R + VEH animals, the relaxation of occluded MCA was drastically and significantly reduced. In the I/R + LPS group, the endothelium-dependent was preserved with an amplitude equivalent to that of sham animals (Table 1 and Fig. 2A). Endothelium-independent relaxation to SNP was similar all four groups (Table 1).
Percentage of maximal variation of diameter of middle cerebral artery in sham-operated and ischemia–reperfusion groups
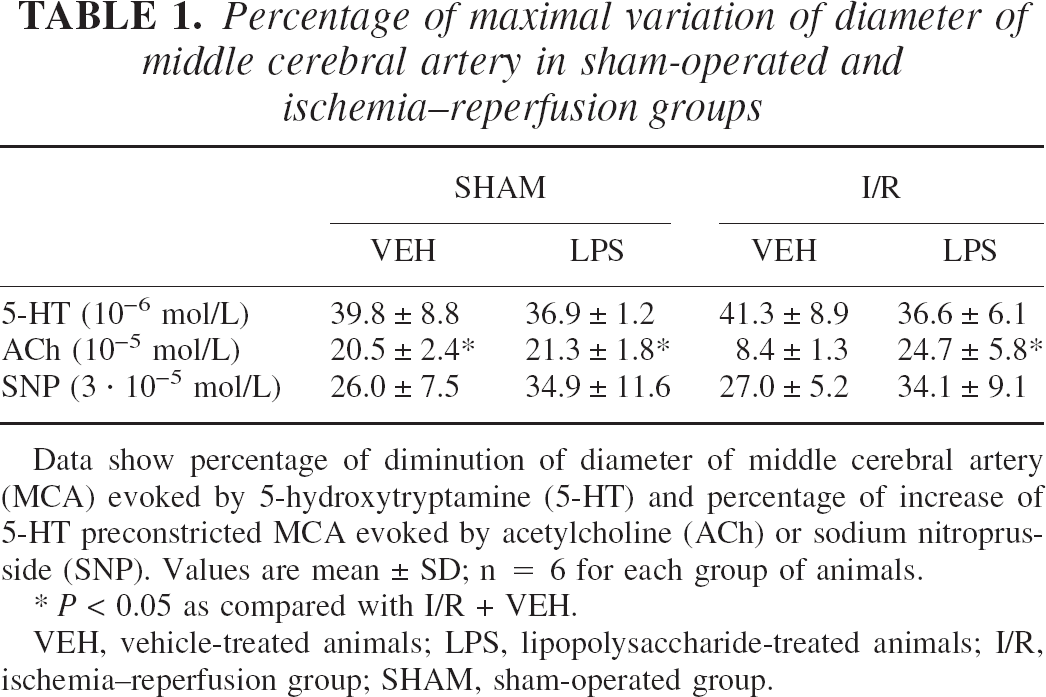
Data show percentage of diminution of diameter of middle cerebral artery (MCA) evoked by 5-hydroxytryptamine (5-HT) and percentage of increase of 5-HT preconstricted MCA evoked by acetylcholine (ACh) or sodium nitroprus-side (SNP). Values are mean ± SD; n = 6 for each group of animals.
P < 0.05 as compared with I/R + VEH.
VEH, vehicle-treated animals; LPS, lipopolysaccharide-treated animals; I/R, ischemia–reperfusion group; SHAM, sham-operated group.
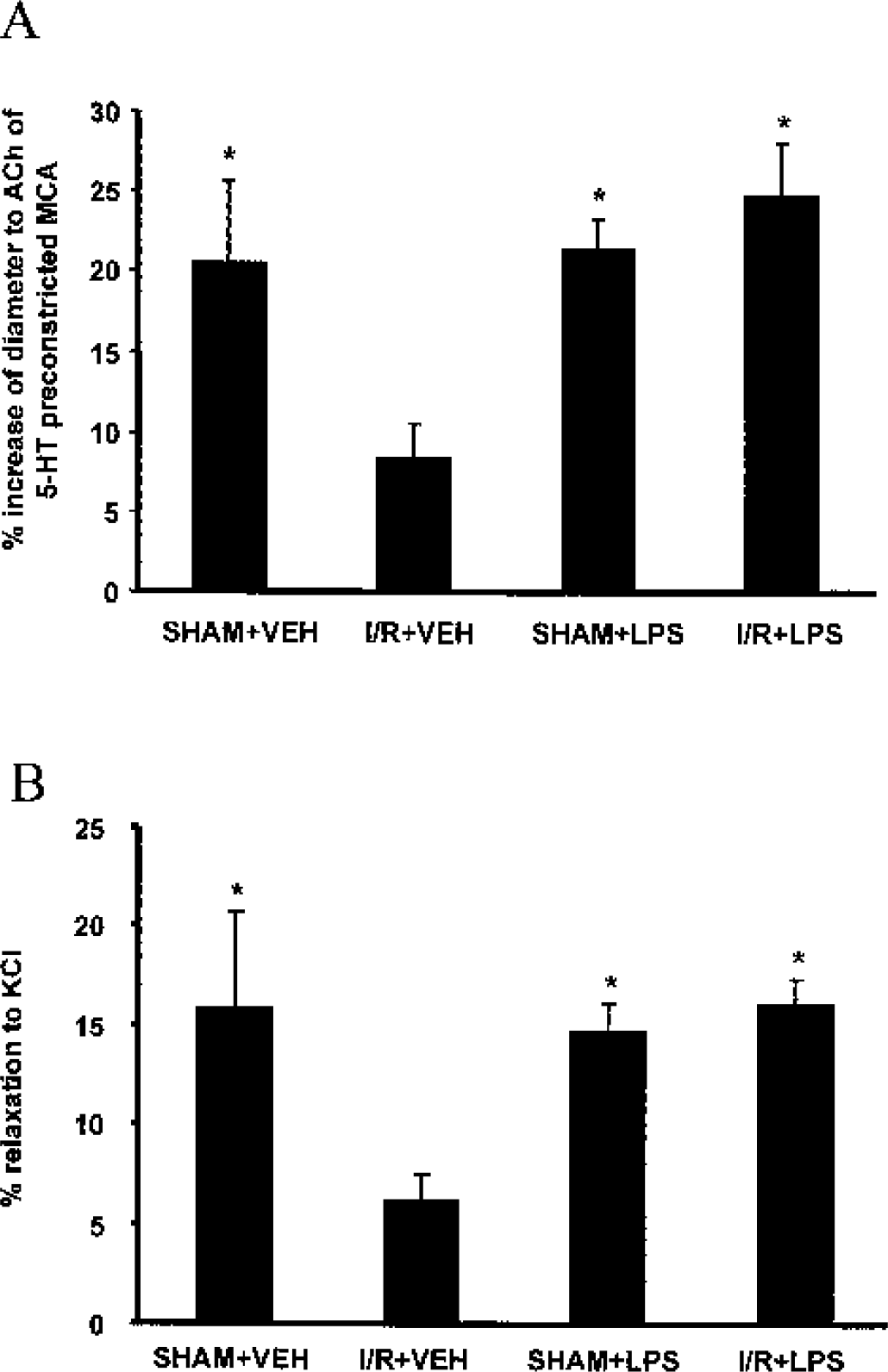
Effect of lipopolysaccharide (LPS) on endothelium-dependent and K+ inward rectifier 2.1–dependent vasorelaxation.
Lipopolysaccharide effects on smooth muscle KIR 2.1–dependent vasoreactivity
Smooth muscle cell-dependent relaxation of MCA was induced by application of 15-mmol/L KCl in the different groups of rats. In SHAM + VEH and SHAM + LPS animals, a relaxation of 15.80% ± 4.99% (n = 6) and 14.71% ± 1.64% (n = 6) was respectively obtained. In I/R + VEH animals, the relaxation of the occluded MCA was significantly reduced to 6.15% ± 1.45% (n = 6; P < 0.01). In the I/R + LPS group, the KCl-induced relaxation of occluded MCA (16.08% ± 1.49% n = 6) was not significantly different from relaxation observed in the MCAs of untreated rats (Fig. 2B)
Lipopolysaccharide effects on KIR 2.1 current density
The transmembrane currents were measured on the freshly isolated VSMC from MCA of the different groups of rats. The capacitance values were not different among groups: 22.07 ± 1.88 pF (n = 8 cells obtained from 5 rats) for SHAM + VEH cells, 21.24 ± 2.01 pF (n = 8 cells obtained from 5 rats) for I/R + VEH cells, 21.20 ± 3.66 pF (n = 7 cells obtained from 4 rats) for SHAM + LPS cells and 21.78 ± 2.9 pF (n = 10 cells obtained from 6 rats) for I/R + LPS cells. The difference between the transmembrane current and the current recorded in presence of 0.5-mmol/L BaCl2 (a specific inhibitor of KIR 2.1) unmasked the proper KIR2.1 component with its characteristic inward rectification. The mean KIR 2.1 current densities (pA/pF) were plotted against potentials (mV) (Fig. 3A). There was a drastic and significant reduction of the amplitude of KIR 2.1 density measured in I/R + VEH MCA cells compared to the superimposed KIR 2.1 densities of SHAM + VEH and I/R + LPS cells. For example, at −135 mV, the potential at which the maximal alteration of KIR 2.1 was observed, the KIR 2.1 density was similar in SHAM + VEH cells (–1.24 ± 0.44 pA/pF, n = 7) and SHAM + LPS cells (–1.50 ± 0.60 pA/pF, n = 7) but fell to −0.49 ± 0.21 pA/pF (n = 8) in I/R + VEH cells (P < 0.05). The previous administration of LPS 72 h before ischemia–reperfusion allowed a significant prevention of the KIR 2.1 alteration (–1.11 ± 0.35 pA/pF, in I/R + LPS cells n = 8, P < 0.01 vs I/R + VEH cells) (Fig. 3 B).
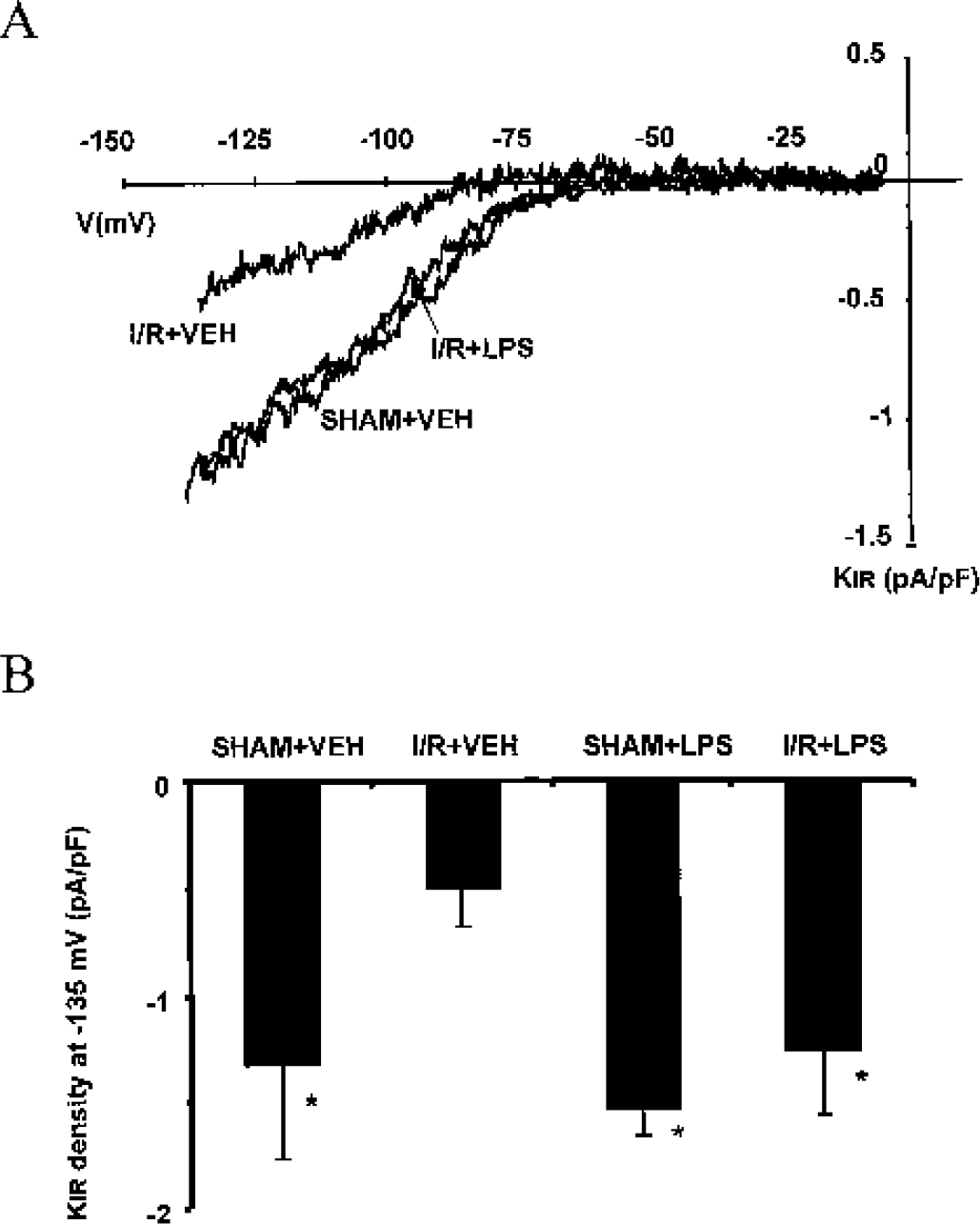
Effect of lipopolysaccharide (LPS) on KIR 2.1 current densities.
DISCUSSION
The present study shows that a low dose of LPS administered 72 h before cerebral ischemia–reperfusion, in addition to its delayed neuroprotective effect, prevents in a delayed manner postischemic cerebrovascular abnormalities. These vascular alterations after the ischemia–reperfusion process concern both the endothelium and smooth muscle layers studied by two different methodologic approaches. Lipopolysaccharide administration prevents the postischemic endothelial dysfunction characterized by an alteration of acetylcholine-dependent vasorelaxation. The ischemia–reperfusion–induced smooth muscle dysfunction, with an impairment in KIR 2.1 current and its related relaxing function, is prevented in animals treated by LPS
The endothelial dysfunction after ischemia–reperfusion process is predominantly the result of reperfusion because there is only a weak decrease in endothelium-dependent relaxation after occlusion alone (Cipolla et al., 1997). The oxidative stress generated by reperfusion could be responsible for endothelial dysfunction occurring in the course of cerebral ischemia (Cai and Harrisson, 2000; Cook and Vollrath, 1995). It has been previously shown that xanthine oxidase–induced superoxide causes reoxygenation injury of ischemic cerebral endothelial cells (Beetsch et al., 1998; Mason et al., 2000) and that there is a decrease in NO bioavailability due to its accelerated degradation by ROS (Cai and Harrison, 2000). In contrast, a decreased NO release could not be involved because endothelial NO synthase expression is increased in the course of cerebral ischemia (Leker et al., 2001). Another mechanism could involve the activation of polymorphonuclear leukocytes shown in ischemia to aggravate tissue damage and to be correlated to the infarct severity, particularly by way of an endothelial dysfunction (Akopov et al., 1996a, b ) probably related to an ROS release (Matsuo et al., 1995).
The finding that KIR current, measured by vasoreactivity or patch-clamp experiments, was impaired suggests that the ion channel component of smooth muscle–dependent vasoreactivity is altered by ischemia–reperfusion. Oxidative stress most likely plays a key role since ROSs are able to induce modification in ion transport pathways (Kourie, 1998). These vascular abnormalities could contribute to extend cerebral lesions as suggested by a significant correlation of impairment of KIR channel density in the occluded artery with the severity of brain injury and brain edema (Bastide et al., 1999).
We evidence here, for the first time, that a low dose of LPS induces a delayed vascular protective effect in parallel to the previously reported delayed neuroprotective effect (Bordet et al., 2000; Tasaki et al., 1997). These data are in accordance with recently reported results showing that ischemic preconditioning can significantly attenuate ischemia-induced brain edema and cerebrovascular injury in the rat (Masada et al., 2001). These results also evoke the delayed endothelial protective effects of monophosphoryl lipid A after myocardial ischemia–reperfusion in rats (Richard et al., 1999) and could explain the LPS-induced resistance associated with a preservation of microvascular perfusion reported by Dawson et al. (1999).
Several mechanisms could underlie these LPS vascular protective effects. A weakening of ROS-induced deleterious effects on vascular wall and of ROS-induced NO degradation, as well as a reduction of activation and adhesion of polymorphonuclear leukocytes as shown in myocardial ischemia (Eising et al., 1996), could be involved. The role of the antioxidant effect of LPS in vascular protection remains elusive because there is no increase in SOD vascular activity (Bastide et al., 2001) and an increase in SOD brain activity (Bordet et al., 2000), but the involvement of a LPS-induced extracellular SOD overactivity cannot be excluded. Moreover, LPS-induced NO release could also contribute to vascular protective effect by inactivation of anion superoxide, the main ROS involved in KIR 2.1 current impairment. Regarding the role of leukocyte activation in the development of KIR 2.1 alteration, it has been shown that it could contribute directly or indirectly to impair vascular smooth muscle function (Ricevuti, 1997).
In parallel to these mechanisms, LPS induced changes in endothelium NO pathway that could also contribute to its vascular protective effect during cerebral ischemia. In myocardium, postischemic endothelial dysfunction is prevented by acute administration of acetylcholine or NO donors, suggesting that a direct or indirect NO pathway is able to restore normal endothelial function. In brain, administration of NO donors prevented decrease in endothelial NO synthase expression in course of subarachnoid hemorrhage, contributing to the endothelial dysfunction and vasospasm (Richard et al., 1995). The beneficial effect of LPS on the cerebral vasculature during ischemia/reperfusion could be related to its ability to increase NO synthase type III in cerebral blood vessel (Puisieux et al., 2000). Moreover, we have reported that this same low dose of LPS also induces a delayed enhanced NO-mediated relaxation in rat peripheral arteries (Pu et al., 1999). The slight LPS-induced increase in relaxing response to acetylcholine observed in the MCA in absence of occlusion could also support this hypothesis. The contribution of NO pathway in KIR 2.1 regulation remains unknown because NO increase has not yet been shown to attenuate the decrease in KIR 2.1 density. Nevertheless, a NO-dependent mechanism has recently been evidenced to be upregulated in the K+-sensitive vasodilation in spontaneously hypertensive rats (Chrissobolis et al., 2002). Direct LPS application on cerebral arteries has been previously found to induce activation of other potassium channel, (e.g., Ca2+-activated K+ channel) by a NO-dependent mechanism (Hoang and Mathers, 1998).
Beyond mechanisms of vascular protection, the question of the relation between neuroprotection and vascular protection remains elusive. The parallelism between these two kinds of protective effects could support the hypothesis of a link between them, particularly experimental arguments suggesting that impairment of smooth muscle–dependent vasorelaxation in occluded MCA contributes to the extent of cerebral ischemia or edema (Bastide et al., 1999) and that loss of microvascular integrin expression during focal brain ischemia reflects neuron injury (Tagaya et al., 2001). These data suggest that a cerebral vascular wall could be a new pharmacological target in stroke and that pharmacological agents mimicking the delayed protective effect of LPS would induce a prophylactic neuroprotection. Moreover, such a vascular protection could be beneficial in some clinical conditions as a means of preventing hemorrhagic complications of thrombolysis, in which vascular dysfunction has been involved (Del Zoppo and Hallenbeck, 2000; Petty and Wettstein, 2001).
Footnotes
Acknowledgments:
The authors thank Mrs. S. Duriez for secretarial assistance.