
Abstract
The authors report that cholecystokinin (CCK), via its subtype 2 receptor (CCK2R) located presynaptically on cerebral arteries, mediates the release of nitric oxide (NO), which induces vasodilatation. Whereas CCK octapeptide and its fragment CCK tetrapeptide (CCK-4) lack a direct effect on the smooth muscle of pial vessels, the authors showed that both CCK peptides modulate the neurogenic responses in bovine cerebral arteries. The neurogenic vasodilatation induced by CCK-4 was blocked by the CCK2R antagonist, L-365,260, and antagonized by neuronal NO synthase (nNOS) inhibitors, but was independent of the endothelium. In whole-mount arteries, CCK2Rs were detected in nerve fibers and colocalized with nNOS and synaptophysin. The findings provide, for the first time, a neural mechanism by which CCK may increase cerebral blood flow.
Cholecystokinin (CCK) is one of the most abundant neuropeptides detected in brain of several mammalian species (Rehfeld, 1978; Vanderhaeghen et al., 1975). C-terminal fragments of the gastrointestinal hormone CCK are synthesized in the central nervous system (CNS), where they act as neurotransmitters and neuromodulators. Cholecystokinin-related peptides interact with two different receptors, the CCK1R, also called “peripheral,” and the CCK2R, which is the most abundant form in the CNS. The C-terminal tetrapeptide of CCK, called CCK-4, is considered a specific CCK2R agonist (Noble et al., 1999).
Cholecystokinin-like immunoreactivity has been found in neurons closely associated with capillary walls and small blood vessels in rat and monkey cerebral cortex (Hendry et al., 1983). Although vascular effects of CCK-related peptides have been reported in peripheric organs (Thulin, 1973; Thulin and Olsson, 1973; Obara et al., 1989), these peptides lack a direct effect on brain blood vessels (McCulloch and Kelly, 1984).
Clinical data suggest, however, that CCK may elicit vasodilatory effects on cerebral arteries. Cholecystokinin-4, as occurs with lactate (Nutt and Lawson, 1992) or CO2 (Fyer et al., 1987; Griez et al., 1987), induces panic attacks in humans (Bradwejn et al., 1990, 1991, 1992; Javanmard et al., 1999). The mechanism underlying the panicogenic action of these agents is not known, but increases of blood flow in particular brain areas are detected almost immediately after their experimental administration (Bradwejn et al., 1990, 1991, 1992; Nutt and Lawson, 1992; Javanmard et al., 1999). In addition, symptoms of panic attacks induced by CCK-4 are detected almost immediately after bolus administration of the peptide, as also occur with changes in cerebral blood flow (CBF) (Benkelfat et al., 1995). Whereas lactate and CO2 are well-known metabolic signals evoking vasodilatation of cerebral arteries, no vascular effects of CCK-related peptides have been previously reported in brain vessels.
Our purpose has been to characterize an eventual vasodilatory effect of CCK on cerebral vessels. In this study we tested whether CCK-related peptides exerted a neurogenic action on bovine pial vessels, characterized the mechanism of their vascular action, and localized CCKRs.
MATERIALS AND METHODS
Bovine brains were obtained from a local slaughterhouse and transported to the laboratory in phosphate-buffered saline solution (PBS) at 4°C. A first-order branch of the anterior cerebral artery was isolated for functional experiments and for biochemical and immunohistochemical determinations. Chemicals were purchased from Sigma (St. Louis, MO, U.S.A.). L-365,260 was a gift of Merck Sharp and Dohme.
Isometric tension recordings
Experiments were performed as previously described (González et al., 1997). Briefly, arterial rings 4 mm in length were suspended on two intraluminal parallel wires, introduced in an organ bath containing a physiologic solution with the following composition: 115 mmol/L NaCl, 4.6 mmol/L KCl, 2.5 mmol/L CaCl2, 25 mmol/L NaHCO3, 1.2 mmol/L KH2PO4, 1.2 mmol/L MgSO4, 0.01 mmol/L ethylene diamine tetraacetic acid (EDTA), and 11 mmol/L glucose at 37°C, and 3 μmol/L dexamethasone to avoid induction of inducible nitric oxide synthase (NOS), and connected to a Piodem strain gauge for isometric tension recording. All segments were given an optimal resting tension of 1 g that was readjusted every 15 minutes during a 90-minute equilibration period. Before starting the experiment, the presence of the endothelium was functionally confirmed by adding bradykinin (10 μmol/L) to prostaglandin F2 (PGF2; 10 μmol/L)-contracted rings. Segments were subsequently washed for another 20-minute equilibration period. To obtain endothelial-free segments, this layer was removed by gently introducing a cotton thread through the vascular lumen, taking care not to damage the adventitial layer. Only rings where the relaxation to bradykinin was abolished were considered as endothelium free.
Responses to transmural nerve stimulation (TNS) were examined to investigate the contraction (200 mA, 0.2 millisecond, 0.5 to 8 Hz). Transmural nerve stimulation–induced relaxation was determined in PGF2-precontracted rings (200 mA, 0.2 millisecond, 1 Hz) (González and Estrada, 1991). We used two parallel platinum electrodes, one at each side of the vessel, connected to a CS-20 stimulator (Cibertec, Madrid, Spain). The neurogenic nature of the stimuli was demonstrated because TNS responses were blocked by tetrodotoxin (1 to 10 μmol/L). The interval between consecutive stimuli was 10 minutes. For relaxation experiments TNS (200 mA, 0.2 millisecond, 1 Hz) always was performed in presence of 5 μmol/L indomethacin, 1 μmol/L phentolamine and 1 μmol/L propranolol, and 0.1 μmol/L atropine to prevent any effect mediated by prostaglandins, noradrenaline, or acetylcholine, respectively (González and Estrada, 1991). Two reproducible responses to TNS were obtained before starting experiments. Inhibitors or antagonists were added to the organ bath 20 minutes before next TNS. Sodium nitroprusside (10 μmol/L) was added at the end of each experiment to obtain the maximal relaxation value. Relaxation induced by 1 Hz was considered 100% (control). Responses obtained in presence of CCK-related peptides as well as inhibitors or antagonists are expressed as percentage of this control relaxation.
Western blotting for CCK2 receptors
Western blots were performed as previously described (Mercer et al., 2000). Briefly, pial cerebral arteries, cerebral rat cortex (positive control), and rat aorta (negative control) were homogenized in 0.1 mmol/L Tris and 5 mmol/L EDTA buffer (pH 6.8) containing 4% sodium dodecyl sulfate and protease inhibitors and then subjected to low-speed centrifugation (2,000g × 5 minutes) to separate cellular debris. Proteins (approximately 1.5 to 2 μg/μL) were separated using sodium dodecyl sulfate–polyacrylamide gel electrophoresis by running them on 10% polyacrylamide gels at 80 to 100 mV for 1 to 2 hours, along with molecular weight markers (10 μL/well), in running buffer (250 mmol/L Tris-HCl, 1.92 mol/L glycine, 0.1% SDS), until the dye front reached the bottom of the gel. Protein bands were transferred to polyvinyl difluoride membranes (Bio Rad, Hercules, CA, U.S.A.) using 100 mV for 2 hours using a Bio-Rad semi-dry Transblot apparatus in transfer buffer (250 mmol/L Tris-HCl, 1,920 mmol/L glycine) at room temperature. The membranes were then washed briefly with Tris-buffered saline (TBS; 50 mmol/L Tris-HCl, 0.95 mol/L NaCl), pH 7.6, and blocked in 5% bovine serum albumin with TBS for 1 hour at room temperature to prevent nonspecific binding of the antibodies to the membranes. After blocking, membranes were washed 3 times with TBS for 15 minutes, then transferred to heat-sealed bags and incubated with primary antibody directed against amino acids 258 to 268 of the CCK2-receptor sequence (Wank et al., 1992) in 1% bovine serum albumin in TBS overnight at 4°C on the rocking plate. The next day, membranes were washed 3 times for 15 minutes in TBS, incubated in horseradish peroxidase–conjugated antirabbit antibody (1:400; Santa Cruz Biochemicals, Santa Cruz, CA, U.S.A.) in sealed bags for 1 to 2 hours and then washed in TBS 3 times. Product was visualized with ECL Western blotting from Amersham Pharmacia Biotech Europe (Freiburg, Germany) by exposure to film. The images were scanned for computer analysis.
Immunohistochemistry
Immunohistochemistry was performed as previously described (González et al., 1997). Briefly, the anterior cerebral artery and its branches were dissected, rinsed, and perfused with PBS, and immediately immersed in 4% paraformaldehyde in 0.1 mol/L phosphate buffer, pH 7.4 at 4°C. The vessels were washed with PBS and incubated overnight at room temperature with a mouse antiserum against synaptophysin (1:200; Sigma), a specific marker of synaptic vesicles (Sudhof et al., 1987), a rabbit antiserum against CCK2R (1:400; Mercer et al., 2000) and with a mouse antiserum against neuronal NOS (nNOS; 1:400; Santa Cruz Biochemicals) diluted in PBS. Arteries were then washed in PBS and incubated with a mixture of Alexa 488-conjugated goat antiserum against rabbit (1:100; Molecular Probes, Eugene, OR, U.S.A.) and a Cy3-conjugated goat antiserum against mouse (1:100; Sigma) for 1 hour at room temperature. Some segments were incubated for 45 minutes in a PBS solution containing 0.1 μmol/L propidium iodide to visualize nuclei of the different vascular layers. Cholecystokinin immunostaining was done with a rabbit antiserum against CCK-8 (1:200; Sigma), followed by incubation with Alexa 488 (1:100; Molecular Probes). Some arterial segments were incubated with Hoechst 33258 (1:200; Molecular Probes) to visualize cell nuclei (Arribas et al., 1997; Moser et al., 1975). The arteries were mounted on slides for visualization with a laser scanning confocal microscope (model MRC 1024; Bio-Rad) coupled to a Nikon microscope with a ×20 air objective (numerical aperture 0.45). Propidium iodide staining of all vascular cells was visualized at 488 nm (excitation wavelength) and 615 nm (emission wavelength).
Statistical analysis
Data are expressed as means ± SEM. Statistical comparison between groups was made by using 1-way or 2-way ANOVA. Individual comparisons between groups receiving either CCK peptides or CCK receptor antagonists or NOS inhibitors were analyzed by the Dunnett test.
RESULTS
Both cholecystokinin-8 and cholecystokinin-4 modulate neurogenic responses in cerebral blood vessels
Cholecystokinin-8 (1 nmol/L to 1 mmol/L) when applied to arterial rings failed to elicit either contractile or relaxant responses (data not shown), in agreement with data obtained in anesthetized cats by other authors (McCulloch and Kelly, 1984).
We then tested the hypothesis that CCK might possess a neurogenic vasoactive action by investigating the effect of exogenous CCK-8 in arteries subjected to TNS. Transmural nerve stimulation elicits the release of both vasoconstrictor and vasodilator neurotransmitters from nerve endings innervating pial arteries (Donald and Shepherd, 1980). Transmural nerve stimulation was given at frequencies leading only to neurogenic responses, since they were abolished by the fast sodium channel blocker tetrodotoxin (1 to 10 μmol/L, data not shown) (González and Estrada, 1991; González et al., 1997). Initially, we characterized the effect of CCK-8 on the contraction due to depolarization of perivascular nerves induced by TNS. Transmural nerve stimulation (0.5–8 Hz) induced frequency-dependent contractions that were significantly reduced by preincubation of vessels with 0.1 μmol/L CCK-8 (Figs. 1A and 1B). The same effect was observed by using the selective CCK2R agonist, CCK-4. Cholecystokinin-4 (0.1 μmol/L) elicited an inhibition of TNS-induced contractions, which was more pronounced than that of CCK-8 at the same concentration (Fig. 1A). The effect of CCK-8 was completely reversed by the CCK2R antagonist, L-365,260 (0.1 μmol/L; Fig. 1C), confirming the involvement of CCK2Rs in this response. L-365,260 had no effect by itself on TNS-induced responses (data not shown).
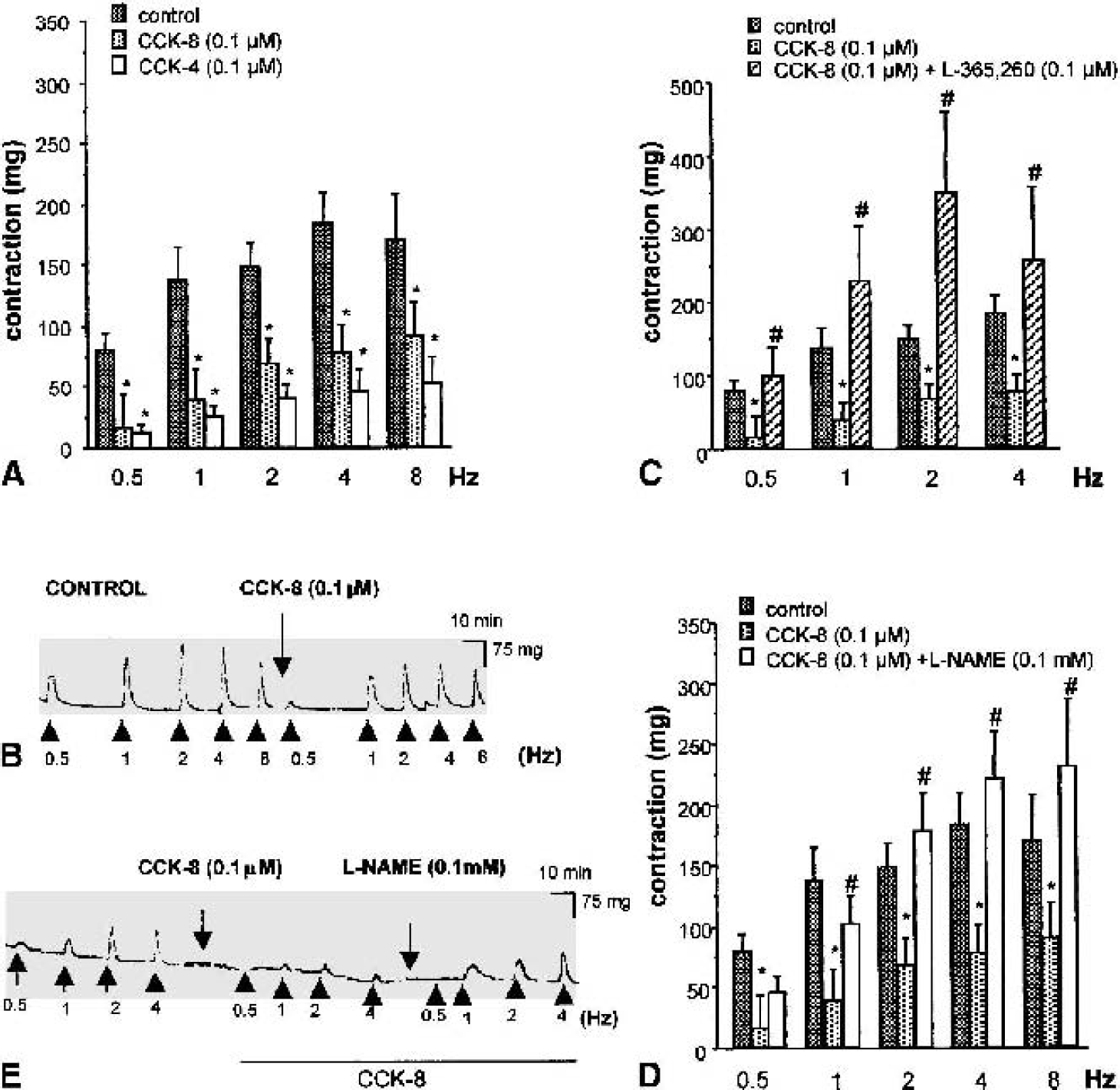
Effect of cholecystokinin (CCK)-related peptides on transmural nerve stimulation (TNS)–induced contraction in bovine anterior cerebral arteries.
Neurogenic vasodilation induced by cholecystokinin-4 is mediated by neuronal nitric oxide
Because TNS induces the release of both vasoconstrictor and vasodilator neurotransmitters, the effect of CCK peptides might be theoretically due to either an enhanced release of dilator or a diminished release of contractile mediators. Indeed, bovine pial arteries are endowed with nitrergic nerves whose stimulation induces a relaxation of arterial segments in vitro (González and Estrada, 1991; González et al., 1997; Yoshida et al., 1993) and an increase in CBF in vivo (Seylaz et al., 1988; Suzuki et al., 1990). Thus, we investigated a possible interaction between CCK and nitrergic mechanisms. Preincubation of the vessels with NG-nitro-l-arginine methyl ester (l-NAME; 0.1 mmol/L), a nonselective NOS inhibitor, completely reversed the inhibitory effect of CCK-8 on TNS-induced contraction (Figs. 1D and 1E). l-NAME alone had no effect on the response elicited by 0.5- and 1-Hz TNS, but significantly increased responses induced by higher frequencies (data not shown).
In a second series of experiments, the effect of CCK-4 was studied in arteries precontracted with 1 μmol/L PGF2. Cholecystokinin-4 lacked a direct effect on these vessels. Precontracted rings were then submitted to TNS in presence of indomethacin, phentolamine, propranolol, and atropine. These experimental conditions allowed the isolation of the vasodilator component of TNS and prevented any effect mediated by prostaglandins, noradrenaline, or acetylcholine. Under these conditions, CCK-4 (10 pmol/L to 0.1 μmol/L) enhanced the relaxation induced by 1-Hz TNS in a concentration-dependent manner, and the enhancement was already significant at the concentration of 1 nmol/L (Fig. 2A). The effect of CCK-4 was blocked by l-NAME (0.1 mmol/L; Fig. 2B), as well as by the specific inhibitor of the nNOS, S-methyl-l-thiocitrulline (SMTC; 0.5 μmol/L; Figs. 2B and 2C) (Furfine et al., 1994). The effect of SMTC was fully reversed by l-arginine, the natural substrate of nNOS (Fig. 2C). The concentration of SMTC used in this experiment is selective for nNOS because it did not modify endothelium-dependent relaxation in response to bradykinin (data not shown). Nonetheless, a possible endothelial source of NO was excluded because removal of endothelium did not modify the concentration-dependent relaxation induced by CCK-4 to 1-Hz TNS (Fig. 2D).
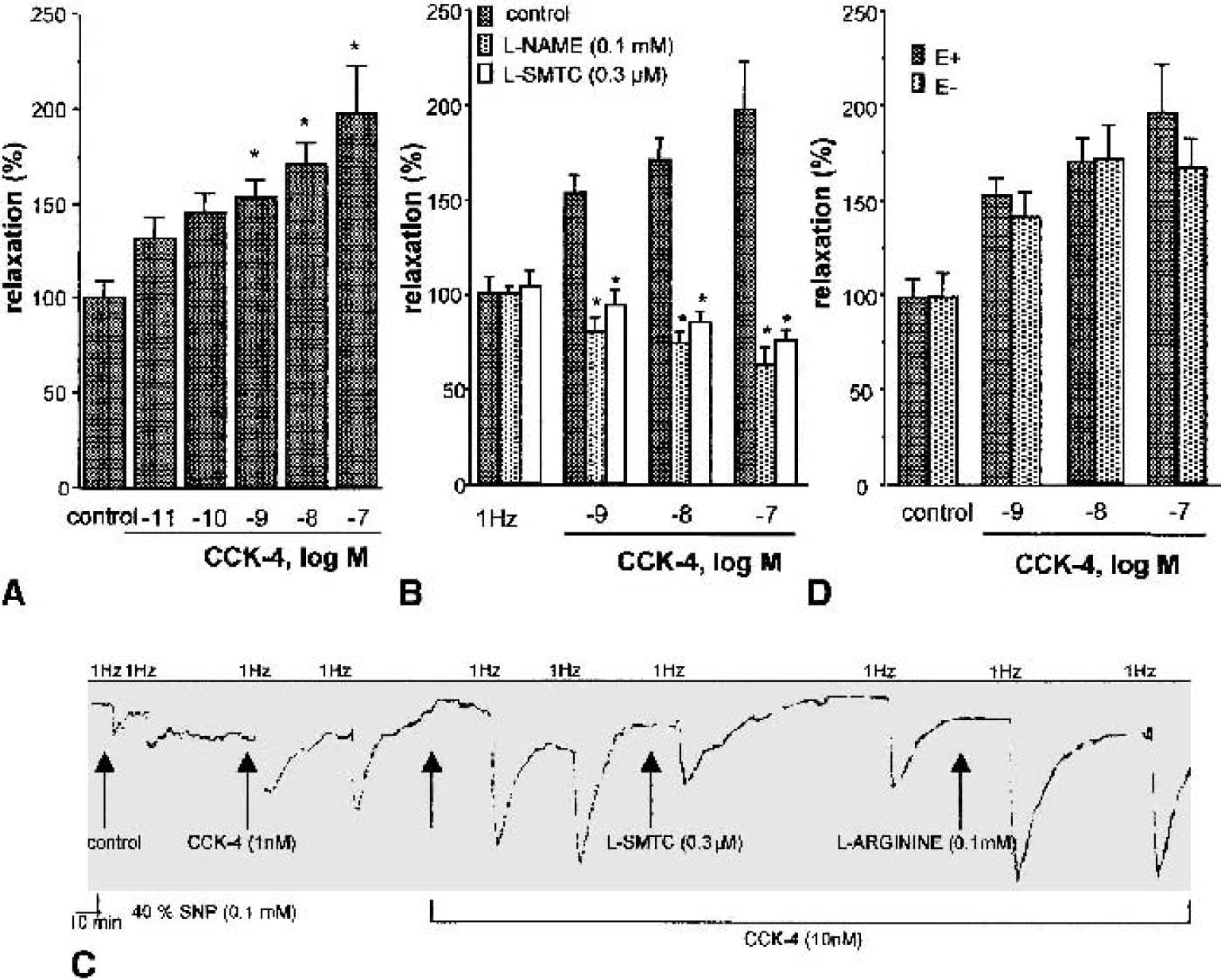
Effect of cholecystokinin (CCK)-4 on 1-Hz transmural nerve stimulation (TNS)–induced relaxation in precontracted bovine anterior cerebral arteries.
Pial cerebral arteries have presynaptic CCK2Rs colocalized with neuronal nitric oxide synthase
In further studies, we sought to determine the presence and localization of CCK2Rs in pial arteries by using both Western blot analysis and immunohistochemistry, respectively. Western blotting was performed in homogenized pial arteries using a selective CCK2R antiserum (Mercer et al., 2000). A major band at 60 kd, compatible with the molecular weight of the CCK2R (Vanderhaeghen et al., 1975) (Fig. 3A), was detected in pial arteries as well as in cerebral rat cortex (positive control), which contains a high density of CCK2Rs, but not in rat aorta (negative control), which lacks CCK2Rs and is a noninnervated vessel.
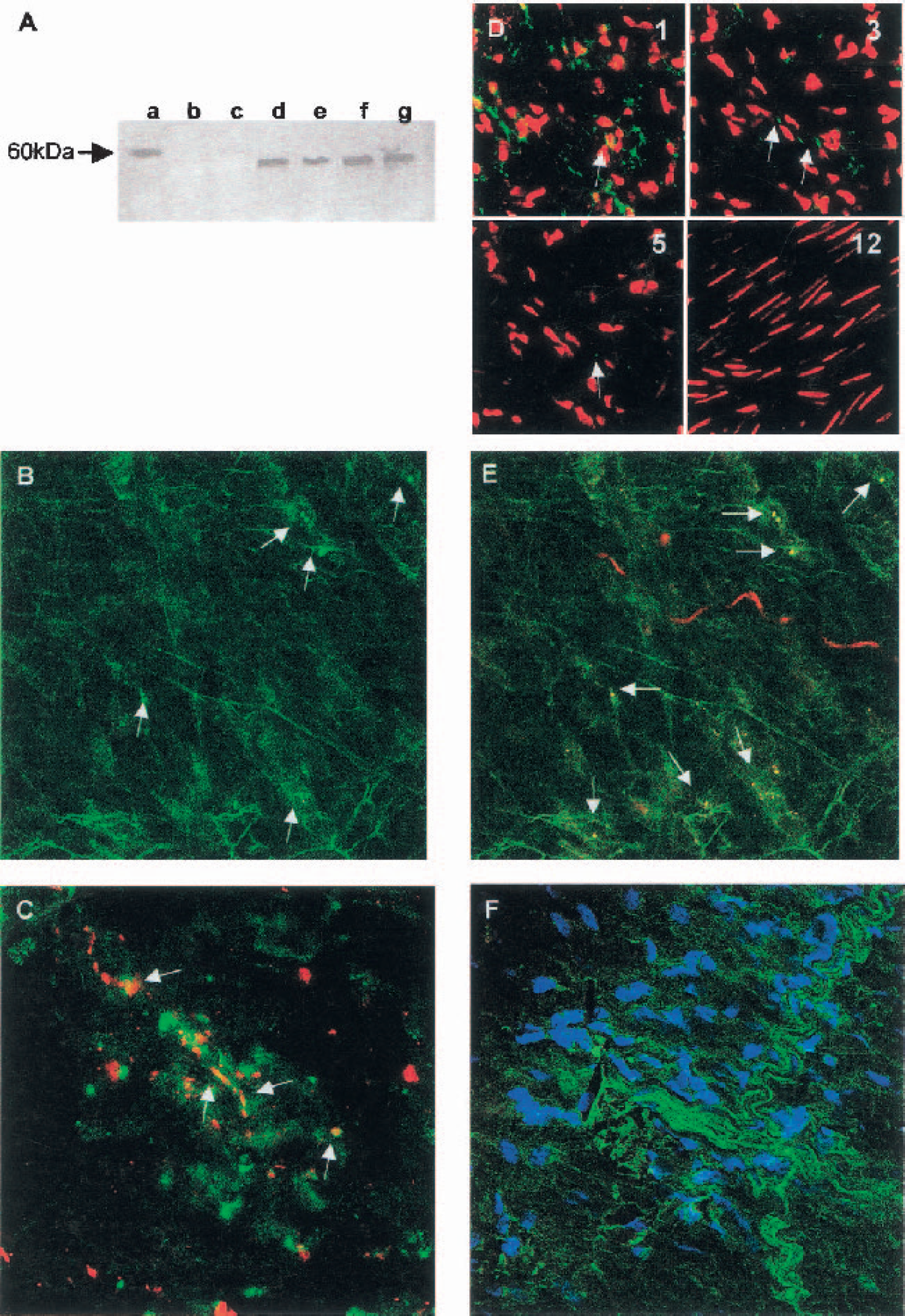
Having demonstrated the presence of CCK2Rs in pial arteries, we then examined their location by cofocal microscopy in vessel whole mounts. Immunohistochemistry using the same antiserum employed in the Western blots, showed fluorescent immunoreactivity for CCK2Rs in bovine pial arteries (Fig. 3B). CCK2Rs are present in the same cellular entities as synaptophysin, a specific marker of synaptic vesicles (Sudhof et al., 1987), and in some cases colocalize with it (Fig. 3C). Fluorescence is thus associated with perivascular nerve endings. Because the morphology of the nuclei of vascular layers is different (adventitial nuclei = round, smooth muscle cell = elongated), propidium iodide staining allowed us to identify the layer containing CCK2Rs. Under our conditions, double staining with propidium iodide and CCK2R antiserum showed that CCK2R-like immunoreactivity was restricted to the adventitial layer (Fig. 3D). Positive immunostaining for nNOS was detected, in the same preparation, in nerve endings showing CCK2R immunoreactivity (Fig. 3E).
Cholecystokinin-8 immunoreactivity is detected in perivascular fibers
Perivascular immunoreactivity to CCK-8 was detected in arterial segments. Cholecystokinin immunostaining appears close to cell nuclei, stained with Hoechst 33258, from the adventitial layer, suggesting that CCK appears in perivascular fibers (Fig. 3F).
DISCUSSION
Our findings show that both CCK-4 and CCK-8 are vasoactive substances that enhance TNS-induced neurogenic dilatation of cerebral vessels mediated by presynaptic CCK2Rs through the activation of nNOS.
We first discarded a direct vasoactive effect of CCK peptides in isolated pial bovine arteries, as reported by other authors (McCulloch and Kelly, 1984). This observation was consistent with our histologic results, which showed that CCK2Rs were not present on the vascular medial layer. Nevertheless, the presence of CCK-ergic neurons close to cerebral vessels (this study, Hendry et al., 1983) led us to hypothesize that CCK could, alternatively, modulate neurogenic vascular responses. This hypothesis was confirmed by the observation that CCK peptides reduced contraction and also enhanced dilatation elicited by TNS.
The neurogenic effect of CCK-4 was dose dependent and was evident from concentrations as low as 1 nmol/L. The involvement of CCK2Rs was further confirmed by using the CCK2R antagonist, L-365,260. We showed the presence of CCK2Rs in pial vessels by Western blotting. Confocal microscopy showed that these receptors were located in the adventitial layer. Immunoreactivity for CCK2Rs was also found in the same cellular entities as synaptophysin, a specific marker of synaptic vesicles (Sudhof et al., 1987), indicating that they were located in perivascular nerve endings. To our knowledge, these results are the first evidence of the presence of CCK2Rs in neuronal endings innervating blood vessels. The existence of presynaptic CCK2Rs has been proposed to explain some pharmacological actions of CCK peptides (Lena et al., 1997), but at present CCKRs had never been visualized in nerve endings of any preparation.
The adventitial layer contains a rich innervation that accounts for the subtle regulation of cerebral vessels. Like other vessels, pial arteries are endowed with nitrergic nerve endings whose stimulation induces a relaxation of arterial segments in vitro (González and Estrada, 1991, González et al., 1997; Yoshida et al., 1993) and an increase in CBF in vivo. (Seylaz et al., 1988; Suzuki et al., 1990). Preincubation of the vessels with SMTC, a selective nNOS inhibitor, completely reversed the effect of CCK-4. SMTC exhibits 17-fold greater selectivity for nNOS (IC50 = 300 nmol/L) than for endothelial NOS (IC50 = 5.4 μmol/L) (Furfine et al., 1994). Nevertheless, we tested the selectivity of SMTC in our experimental setting, confirming that the concentration chosen did not affect bradykinin-induced dilatation, which is endothelial NOS–mediated (Vallance et al., 1989). This result showed that the vasodilator effect of CCK-4 was linked to the release of neuronal NO. This functional study is supported by the histologic data showing that nNOS was present in the adventitial layer of pial vessels and colocalized in the same cells with CCK2Rs.
CONCLUSIONS
These data constitute the first demonstration that CCK peptides, by acting on presynaptic CCK2Rs, modulate neurogenic responses in cerebral blood vessels and suggest that CCK could act in the CNS as a vasodilator mediator. From our results we cannot assess the physiologic or pathophysiologic relevance of such an effect of CCK peptides on brain vessels. Our data, however, interpreted in the context of the CCK-4 hypothesis of panic disorder, led us to speculate that the increase of CBF that appears associated with spontaneous and induced panic could be related to the vasodilatory effect of CCK: (1) symptoms of panic attacks induced by CCK-4 are detected almost immediately after bolus administration of small doses (nmol/kg) of the peptide, as also occurs with changes in CBF (Bradwejn et al., 1991, 1992; Benkelfat et al., 1995); (2) lactate and CO2, which induce panic episodes similar to those elicited by CCK-4, are well-known metabolic signals evoking vasodilatation of cerebral arteries; and (3) the increase in regional CBF after CO2 inhalation has been shown to be mediated essentially by NO produced by nNOS (Wang et al., 1995).
In summary, we provide the first demonstration that CCK modulates neurogenic responses in cerebral blood vessels, leading to a vasodilation mediated by NO. Our data represent a conceptual advance in the physiology and pharmacology of CCK that may help to explain actions of CCK that are at present poorly understood.