
Abstract
The cellular and molecular pathways initiated by traumatic brain injury (TBI) may compromise the function and structural integrity of mitochondria, thereby contributing to cerebral metabolic dysfunction and cell death. The extent to which TBI affects regional mitochondrial populations with respect to structure, function, and swelling was assessed 3 hours and 24 hours after lateral fluid—percussion brain injury in the rat. Significantly less mitochondrial protein was isolated from the injured compared with uninjured parietotemporal cortex, whereas comparable yields were obtained from the hippocampus. After injury, cortical and hippocampal tissue ATP concentrations declined significantly to 60% and 40% of control, respectively, in the absence of respiratory deficits in isolated mitochondria. Mitochondria with ultrastructural morphologic damage comprised a significantly greater percent of the population isolated from injured than uninjured brain. As determined by photon correlation spectroscopy, the mean mitochondrial radius decreased significantly in injured cortical populations (361 ± 40 nm at 24 hours) and increased significantly in injured hippocampal populations (442 ± 36 at 3 hours) compared with uninjured populations (Ctx: 418 ± 44; Hipp: 393 ± 24). Calcium-induced deenergized swelling rates of isolated mitochondrial populations were significantly slower in injured compared with uninjured samples, suggesting that injury alters the kinetics of mitochondrial permeability transition (MPT) pore activation. Cyclosporin A (CsA)-insensitive swelling was reduced in the cortex, and CsA-sensitive and CsA-insensitive swelling both were reduced in the hippocampus, demonstrating that regulated MPT pores remain in mitochondria isolated from injured brain. A proposed mitochondrial population model synthesizes these data and suggests that cortical mitochondria may be depleted after TBI, with a physically smaller, MPT-regulated population remaining. Hippocampal mitochondria may sustain damage associated with ballooned membranes and reduced MPT pore calcium sensitivity. The heterogeneous mitochondrial response to TBI may underlie posttraumatic metabolic dysfunction and contribute to the pathophysiology of TBI.
Keywords
Mitochondrial damage may contribute to the neurodegeneration associated with many diseases and disorders by amplifying both apoptotic and necrotic cell death pathways (Green and Kroemer, 1998; Nicholls and Budd, 2000). Bioenergetic failure can recruit mitochondria to execute cell death (Kroemer et al., 1998; Kroemer and Reed, 2000). The activation of the mitochondrial permeability transition (MPT) pore can trigger cell death in a variety of tissues, including brain (Zoratti and Szabo, 1995; Kristal and Dubinsky, 1997; Susin et al., 1998; Friberg et al., 1999; Berman et al., 2000; Kristal et al., 2000). Mitochondrial permeability transition pore activation occurs when matrix calcium concentrations exceed the calcium gating potential, which is modulated by a series of inducers (free radicals, thiol oxidation, pH) and inhibitors (magnesium, ADP, cyclosporin A (CsA) (Halestrap and Davidson, 1990; Bernardi et al., 1999). Activation of the highly regulated, nonspecific MPT pore is characterized by the formation of contact sites between the inner and outer mitochondrial membranes, loss of mitochondrial membrane potential (ΔΨM), cristae disruption, mitochondrial swelling, and release of apoptogenic proteins (Kroemer et al., 1998; Brdiczka et al., 1998). The MPT pore activation in mitochondrial populations may be represented as a continuum between regulated (low-conductance, CsA-sensitive) and unregulated (high-conductance, CsA-insensitive) states (Novgorodov and Gudz, 1996; Gogvadze et al., 2001; He and Lemasters, 2002).
Metabolic disruption is a consequence of traumatic brain injury (TBI) (Yoshino et al., 1991; Moore et al., 2000) and may include a mismatch between glucose metabolism and blood flow (Ginsberg et al., 1997; Moore et al., 2000), reduced cytochrome oxidase activity (Hovda et al., 1991), lower ATP concentration (Signoretti et al., 2001), and increased lactate accumulation (Kawamata et al., 1995). As such, deficits in mitochondrial respiratory coupling are evident after controlled cortical impact (CCI) (Xiong et al., 1997), but not lateral fluid-percussion (FP) (Vink et al., 1990) brain injury in the rat. The structural alterations in mitochondria (e.g., ballooning and membrane destruction) that have been documented after experimental hypoglycemia (Friberg et al., 1998), cerebral ischemia (Brown and Brierley, 1972; Schweiger et al., 1988; Solenski et al., 2002), and TBI (Pettus et al., 1994) are similar to those observed in isolated CNS mitochondria exposed to calcium (Kristian et al., 2000; Brustovetsky et al., 2002).
Increased calcium accumulation (Fineman et al., 1993), free radical generation (Lewen and Hillered, 1998), and caspase-3 activation (see Raghupathi et al., 2000 for review) observed after TBI are ideal conditions for MPT induction (Zoratti and Szabo, 1995; Susin et al., 1998). There is no direct evidence, however, for MPT pore activation after CNS injury. Nonetheless, pharmacologic interventions targeted to reduce MPT pore activation attenuate histopathologic alterations associated with experimental hypoglycemia (Friberg et al., 1998), cerebral ischemia (Matsumoto et al., 1999), and TBI (Scheff and Sullivan, 1999). Furthermore, administration of the calcium channel blocker SNX-111 or CsA preserves mitochondrial structure and function after experimental TBI (Verweij et al., 1997; Okonkwo and Povlishock, 1999; Sullivan et al., 1999). The efficacy of CsA in experimental TBI models, however, may not be mediated via mitochondria, because the MPT-inactive analog FK-506 retains efficacy similar to CsA (Singleton et al., 2001). In experimental cerebral ischemia, however, the neuroprotection provided by the MPT-selective, nonimmunosuppressive N-methyl-val-4-cyclosporin A analog substantiates the importance of mitochondrial preservation after CNS injury (Matsumoto et al., 1999).
In the present study, we hypothesized that the metabolic deficit (Yoshino et al., 1991; Moore et al., 2000) and cell loss evident in the selectively vulnerable parietotemporal cortex and hippocampus after TBI (Dietrich et al., 1994; Hicks et al., 1996) may result from structural and functional damage to mitochondrial populations: specifically, whether brain injury alters the composition of mitochondrial populations to include mitochondria with high-conductance, unregulated MPT pores. In the acute postinjury period after lateral FP brain injury in rats, alterations in mitochondrial yield, in vitro and in vivo function, morphology, size, and in vitro MPT pore activation showed heterogeneous mitochondrial populations in the parietotemporal cortex and hippocampus based on differential sensitivities to injury, calcium, and CsA.
MATERIALS AND METHODS
All animal experiments were approved by both the Animal Use Committee at the Lund University Hospital and the Institutional Animal Care and Use Committee of the University of Pennsylvania. In all studies we adhered to the animal welfare guidelines set forth in the Guide for the Care and Use of Laboratory Animals (National Research Council, National Academy Press, Washington, DC, 1996). Cyclosporin A was obtained from Novartis (Basel, Switzerland), and remaining chemicals were obtained from Sigma-Aldrich (St. Louis, MO, U.S.A.).
Animals, surgery, and fluid percussion injury
A total of 62 male Sprague-Dawley rats (Harlan, Indianapolis, IN, U.S.A.) and 10 male Wistar rats (Moellegard avlslaboratorium, Copenhagen, Denmark), weighing 350 to 400 g, were subjected to anesthesia (sodium pentobarbital, 60 mg/kg, intraperitoneally) and either surgery without injury (sham) or lateral FP brain injury of moderate severity (2.6 to 2.8 atm), as originally described (McIntosh et al., 1989). Briefly, animals were randomized between sham (uninjured) control (n = 22) and injured (n = 50) groups, anesthetized, and placed in a stereotactic frame. The scalp and temporal muscle were reflected and a 5.0-mm craniotomy was made in the skull over the left parietal cortex, between bregma and lambda. A female Luer-Lok fitting was cemented into the open craniotomy and was then used to attach the animal to the FP device. To induce the injury, a hammer was dropped, striking the piston of a saline-filled reservoir, forcing a jet of saline to impact the intact dural surface of the brain, causing a 20-millisecond mechanical deformation. Prior studies in our laboratory have failed to detect alterations in physiologic parameters (blood PO2, PCO2, pH, mean arterial blood pressure, or heart rate) within the first 24 hours after lateral fluid percussion brain injury of moderate severity (Bareyre et al., 1999; Sanderson et al., 1999). Although nine animals died of respiratory depression/apnea within 15 minutes after injury (18% mortality), the duration of postinjury apnea, as a surrogate measure of secondary hypoxia, did not correlate with the results associated with mitochondrial function.
TTC and Nissl staining
Between 30 minutes and 36 hours after injury, injured brains (n = 3 to 5/time point) were rapidly removed and 2-mm coronal sections were incubated in 2% 2,3,5-triphenyltetrazolium chloride (TTC; 0.2 mol/L phosphate buffer, pH 7.4, 37°C) for 6 minutes, 3 minutes per side, while occluded from light. Sections were fixed for 48 hours (10% formalin, 4°C) before digitally imaging the anterior face under coverglass. To evaluate the gross histopathology associated with mitochondrial enzyme dysfunction, 14-μm sections were cut from the 2-mm sections on a cryostat, thaw mounted on gelatin-coated slides, and stained with 0.1% cresyl violet. Digital micrographs of the parietotemporal cortex of the left hemisphere were captured under light microscopy.
Mitochondrial isolation
Somal (nonsynaptosomal) mitochondria were isolated at 3 hours and 24 hours after FP injury or 24 hours after anesthesia and surgery from the parietotemporal cortex and the hippocampus ipsilateral to the craniotomy of individual animals, as previously described (Sims, 1990). Brains were rapidly removed and placed in ice-cold isolation buffer (320 mmol/L sucrose, 2 mmol/L ethyleneglycol tetraacetic acid [EGTA], 10 mmol/L Tris-base, pH 7.4). To reduce the amount of uninjured tissue, the left hemisphere was dissected grossly in ice-cold isolation buffer to segregate the injured parietotemporal cortex (∼100 mg) and the whole hippocampus, including the dorsal and ventral blades of the dentate (∼80 mg). Brain regions from each animal were weighed and then homogenized separately in a 12% Percoll solution in isolation buffer, using 7 strokes of a 4-mL Teflon homogenizer (Kontes, Vineland, NJ, U.S.A.). The homogenate was added onto a discontinuous Percoll gradient (40% and 26%) and centrifuged (8 minutes, 31,000g). The third band, lying on top of the 40% Percoll, was extracted as the mitochondrial fraction, stabilized with 0.5 mg bovine serum albumin (BSA)(Lai and Clark, 1989) and rinsed twice by centrifugation (14 minutes, 17,000g; 5 minutes, 7,300g) in isolation buffer. The final pellet was resuspended in isolation buffer and the protein concentration was determined using the Bio-Rad DC protein assay (Bio-Rad, Richmond, CA, U.S.A.) with BSA standards. Mitochondrial protein yields were expressed as micrograms of mitochondrial protein per milligram of wet tissue.
Mitochondrial respiratory coupling
Oxygen consumption was measured in mitochondria isolated from the cortex and hippocampus 3 hours (n = 4) and 24 hours (n = 5) after injury or 24 hours after sham surgery (n = 4). The final pellet was resuspended in isolation buffer and analyzed in respiration buffer (10 mmol/L Tris-base, 5 mmol/L Tris-phosphate, 50 μmol/L ethylenediamine tetraacetic acid, 100 mmol/L KCl, 75 mmol/L D-mannitol, 25 mmol/L sucrose, 0.2 mg/ml BSA, pH 7.4) maintained at 28°C. The rates of oxygen consumption (nmoles O2 · min−1 · mg−1 of mitochondrial protein) were measured with a Clark-type oxygen electrode in an airtight chamber (Hansatech Instruments, PP Systems, Haverill, MA, U.S.A.) with 5 mmol/L malate and 5 mmol/L glutamate as substrates in the presence (state 3) and absence (state 4) of 140 nmol/L ADP (Sims, 1990). The respiratory control ratio was calculated as the ratio between state 3 and state 4 respiration.
Tissue ATP concentration
Under alkaline conditions, ATP was extracted from frozen biopsies and quantified as previously described (Hylton et al., 1995). At 3 hours (n = 5) or 24 hours (n = 6) after injury or 24 hours after surgery without injury (n = 5), animals were intubated and anesthetized with 2% halothane and 15% O2. A tail artery cannula was used to monitor cessation of blood pressure and heart rate as liquid nitrogen was funneled over the exposed craniotomy. The head was coronally bisected through the center of the craniotomy and immersed in liquid nitrogen. In a glove box cooled to −20°C, 100 to 200 μm of tissue was shaved from the exposed, anterior-facing coronal surface. The sensitivity of the technique permitted selective sampling of the injured parietotemporal cortex and the vulnerable CA3 region of the hippocampus ipsilateral to the site of injury using a squared-off 16G syringe needle. At −20°C, the samples were weighed (1 to 2 mg wet weight) and added to 0.1N NaOH in methanol (100 μL). After 15 minutes at −20°C, the softened slurries were placed on ice, combined with 0.05N NaOH (200 μL) and stored for 15 minutes on ice. After mixing, the extracts were incubated at 95°C for 10 minutes to inactivate tissue enzymes. ATP was assayed by mixing tissue extract (5 μL) with luciferin/luciferase reagent (100 μL; 95 mmol/L Na2HAsO4, 95 mmol/L dithiothreitol, 95 mmol/L EGTA, 950 mmol/L Mg acetate, 0.01% BSA, 0.05 mmol/L luciferin, 0.05 mg/mL luciferase) and measuring light emission in an LKB Wallac 1250 Luminometer (PerkinElmer, Emeryville, CA, U.S.A.). ATP concentration (mmol ATP/kg wet tissue) was calculated against an internal ATP standard solution subjected to the identical extraction procedure.
Mitochondrial ultrastructure
The ultrastructural morphology of mitochondria isolated from the cortex and hippocampus of uninjured control (n = 3) and brain-injured animals at 3 hours (n = 3) and 24 hours (n = 3) after TBI was evaluated using electron microscopy. The final pellet was resuspended in isolation buffer containing 0.85 μmol/L CsA and centrifuged at 15,000g for 45 minutes (2°C) (Friberg et al., 1998). The supernatant fraction was decanted and the pellets were fixed overnight (2.5% glutaraldehyde in cacodylate buffer) at 4°C. Isolated mitochondrial pellets were processed by the Biomedical Imaging Core Laboratory (Department of Pathology and Laboratory Medicine, University of Pennsylvania) for routine thin-section ultrastructural analysis. Randomly selected noncontiguous, nonoverlapping, digitized images of each mitochondrial pellet (24,348x magnification) were captured. To control for variability in isolation yields and pellet density, the first 60 mitochondria in 4 to 5 images per pellet were evaluated by an observer blinded to the source of mitochondria.
Mitochondria were designated as either condensed or damaged. Condensed (healthy) mitochondria were classified by compacted matrix cristae and a continuous outer membrane, regardless of size or shape. In contrast, damaged (swollen) mitochondria were classified primarily by fragmented cristae with evidence of an expanded (ballooned) matrix compartment or disrupted outer membrane. The percentage of damaged mitochondria in the mitochondrial population from each group was calculated. Because of potential osmotic changes induced during fixation, quantitative measurements of mitochondrial dimensions from the electron micrographs were avoided.
Mean radius of isolated mitochondrial populations
Because mitochondria assume a spherical geometry in isolation (Kristal and Dubinsky, 1997), the mean hydrodynamic radius of unfixed, viable mitochondrial populations could be quantified using dynamic light scatter principles for spherical particles used in photon correlation spectroscopy (Glatter et al., 1991). Qualitatively, the translational diffusion coefficient of aqueous particles varies inversely with hydrodynamic radius, such that larger particles have smaller diffusion coefficients. The photon correlation spectroscopy technique takes advantage of the diffusion associated with simple Brownian motion of mitochondria by autocorrelating intensity fluctuations of scattered light (782 nm) as a function of delay time. The hydrodynamic radius of the average scattering particle (mean particle size) is calculated from the diffusion coefficient using the Stokes-Einstein relation. The power of dynamic light scatter lies in the fact that no additional measurements of refractive index or particle concentration are necessary to obtain the mean mitochondrial radius within the population. The mean mitochondrial radius of mitochondrial populations isolated from the cortex and hippocampus of uninjured (n = 5), 3 hour (n = 5) and 24 hour (n = 5) injured animals was acquired. Mitochondria (diluted to empirically yield 200,000 to 500,000 scattered photons per second) were acclimated in isotonic deenergized buffer (150 mmol/L KCl, 20 mmol/L MOPS, 10 mmol/L Tris, 2 mmol/L nitrilotriacetic acid, 0.5 μmol/L rotenone, 0.5 μmol/L antimycin, and 2 μmol/L A23187, pH 7.0) at 26°C for 2 minutes before scattered light intensity data were acquired (20 to 25 measurements, 5-second acquisition time each) and correlated over 120 exponentially distributed delay times (DynaPro-MS/X, Protein Solutions, Lakewood, NJ, U.S.A.). The mean hydrodynamic radius (nanometers) for mitochondrial populations isolated from uninjured and injured brain was solved from the diffusion coefficient of the autocorrelation function.
Mitochondrial permeability transition pore activation in response to calcium challenge and cyclosporin A
Mitochondria were challenged with varying concentrations of exogenous calcium under deenergized conditions either in the absence or presence of the MPT pore inhibitor CsA. The rate of calcium-induced osmotic swelling of mitochondria under deenergized conditions represents the initial response of the pore components to pathologic levels of calcium (without respiration-related compensation of the calcium-induced changes), and provides an indication of MPT pore kinetics (Connern and Halestrap, 1994; Brustovetsky and Dubinsky, 2000a). The calcium-induced swelling rates were measured in cortical and hippocampal mitochondria isolated 3 hours after brain injury (n = 5) or surgery (n = 5). The reduced mitochondrial protein yield at 24 hours after injury prevented swelling from being assessed. The final pellet was resuspended in isolation buffer, and 11.2 ± 0.75 μg of mitochondrial protein was assayed in 1.0 mL of 26°C isotonic deenergized swelling buffer. Oxidative phosphorylation inhibitors (rotenone, antimycin) and a calcium ionophore (A23187) were used to establish deenergized conditions and ensure complete equilibration of calcium ions across the mitochondrial membrane, respectively (Connern and Halestrap, 1994). Mitochondrial swelling was followed on an LS-5B PerkinElmer fluorometer as the decrease in 90° light scatter at 520 nm (Friberg et al., 1999). Baseline light scatter was monitored for 1 to 2 minutes before adding CaCl2 (20, 50, 100, or 200 μmol/L final concentration) and recording reductions in light scatter for 3 to 5 minutes. The total calcium-induced swelling rates (reduction in absorbance of scattered light·min−1-μg−1 mitochondrial protein; arbitrary units) were calculated as the steepest tangential slope over the first 1 to 2 minutes after calcium addition. Separate experiments with 1.0 μmol/L CsA added to the swelling assay buffer, followed by calcium exposure (50 or 100 μmol/L final concentration), were conducted to distinguish CsA-insensitive from CsA-sensitive swelling, and elucidate the mechanism of calcium-induced mitochondrial swelling. Cyclosporin A-insensitive swelling rates were calculated from trials with CsA and subtracted from total swelling rates to obtain CsA-sensitive swelling rates. The nitrilotriacetic acid buffering capacity of calcium was taken into consideration when calculating the final calcium concentrations (Connern and Halestrap, 1994).
Statistical analysis
Data from cortex and hippocampus were analyzed separately, because recent evidence has shown regional differences in terms of mitochondrial function and MPT pore activation in naive uninjured rats (Friberg et al., 1999). Data obtained from the contralateral cortex and hippocampus in terms of TTC, Nissl, ATP, protein yield, respiration, ultrastructure, and calcium sensitivity were not significantly different from data from uninjured control hemispheres, and are not reported for the sake of brevity. Data are presented as means ± SD with statistical differences among groups (sham, 3-hour injured, 24-hour injured) analyzed by one-way (protein yield, respiration, ATP, morphology, and mean radius), two-way (calcium-induced swelling), or three-way (CsA-sensitive swelling) analysis of variance followed by a Newman-Keuls post hoc comparison. A P value less than 0.05 was considered statistically significant.
RESULTS
Mitochondrial enzyme dysfunction precedes cell loss
Preliminary studies using the reduction of TTC, an indicator of mitochondrial succinate dehydrogenase (complex II) enzymatic activity (Lippold, 1982; Perri et al., 1997), were conducted to associate mitochondrial enzyme dysfunction with gross histopathology. The onset of mitochondrial enzyme dysfunction, as indicated by less red formazan product (Lippold, 1982), was detected at 3 hours after injury (Fig. 1A) without an overt loss of tissue (Fig. 1B). Noticeable loss of enzymatic activity in the cortex by 24 hours (Fig. 1C) coincided with the loss of Nissl stain from corresponding TTC sections evident at 24 hours after injury in the parietotemporal cortex (Fig. 1D). By 24 hours after injury, a cavity has yet to form, but reliably forms by 1 week after injury (Hicks et al., 1996; Smith et al., 1997). Consequently, the remaining analyses were conducted at 3 hours and 24 hours after injury, to evaluate mitochondria from tissue with progressively more metabolic dysfunction.
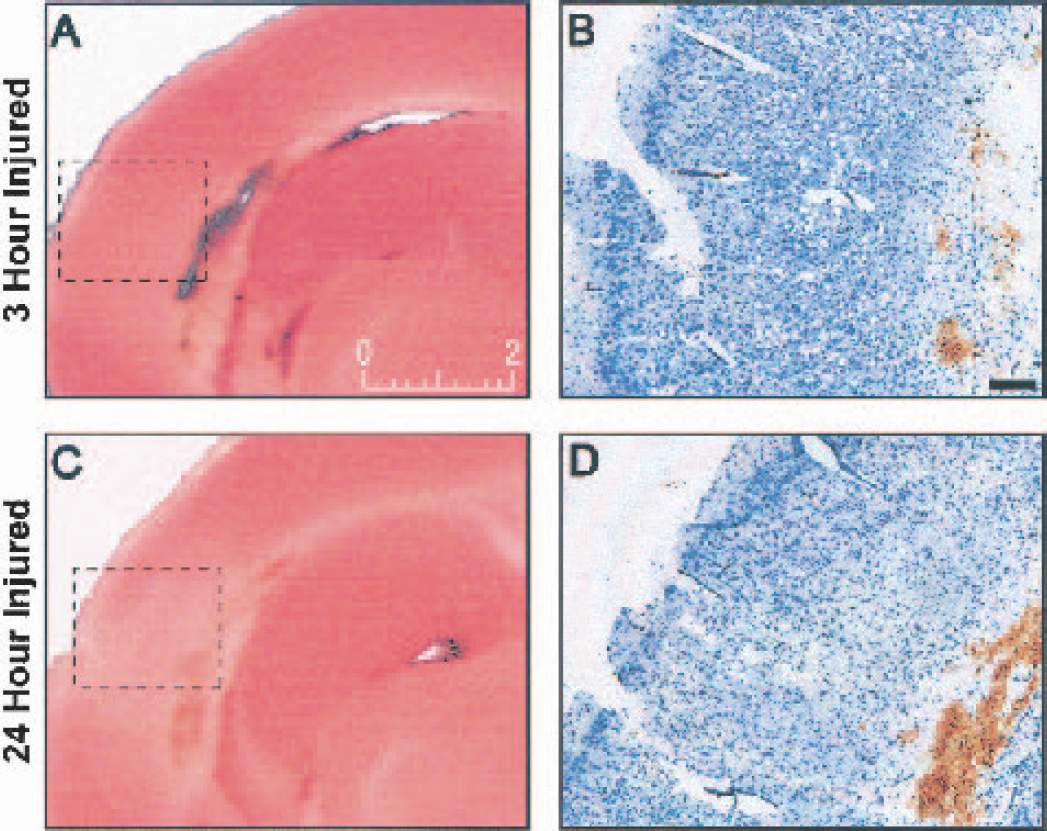
Histochemical evaluation of mitochondrial enzyme dysfunction precedes posttraumatic histologic vulnerability. Three hours
Less mitochondrial protein isolated from injured cortex
The discontinuous Percoll gradient, nonsynaptosomal mitochondria isolation protocol resulted in comparable yields of mitochondrial protein per milligram tissue from the parietotemporal cortex and the hippocampus of the left hemisphere of uninjured control (sham) animals (Table 1). Mitochondrial protein yields from the left parietotemporal cortex were significantly reduced by 46% at 3 hours and by 60% at 24 hours after injury compared with sham (uninjured) control values (P < 0.05). The yields of mitochondrial protein from the left hippocampus of injured animals were not significantly different than those from uninjured animals.
Mitochondrial protein yield, isolated mitochondrial respiratory function, and tissue ATP concentrations for the parietotemporal cortex and hippocampus of rats 24 hours after surgery (sham) and 3 and 24 hours after fluid percussion brain injury
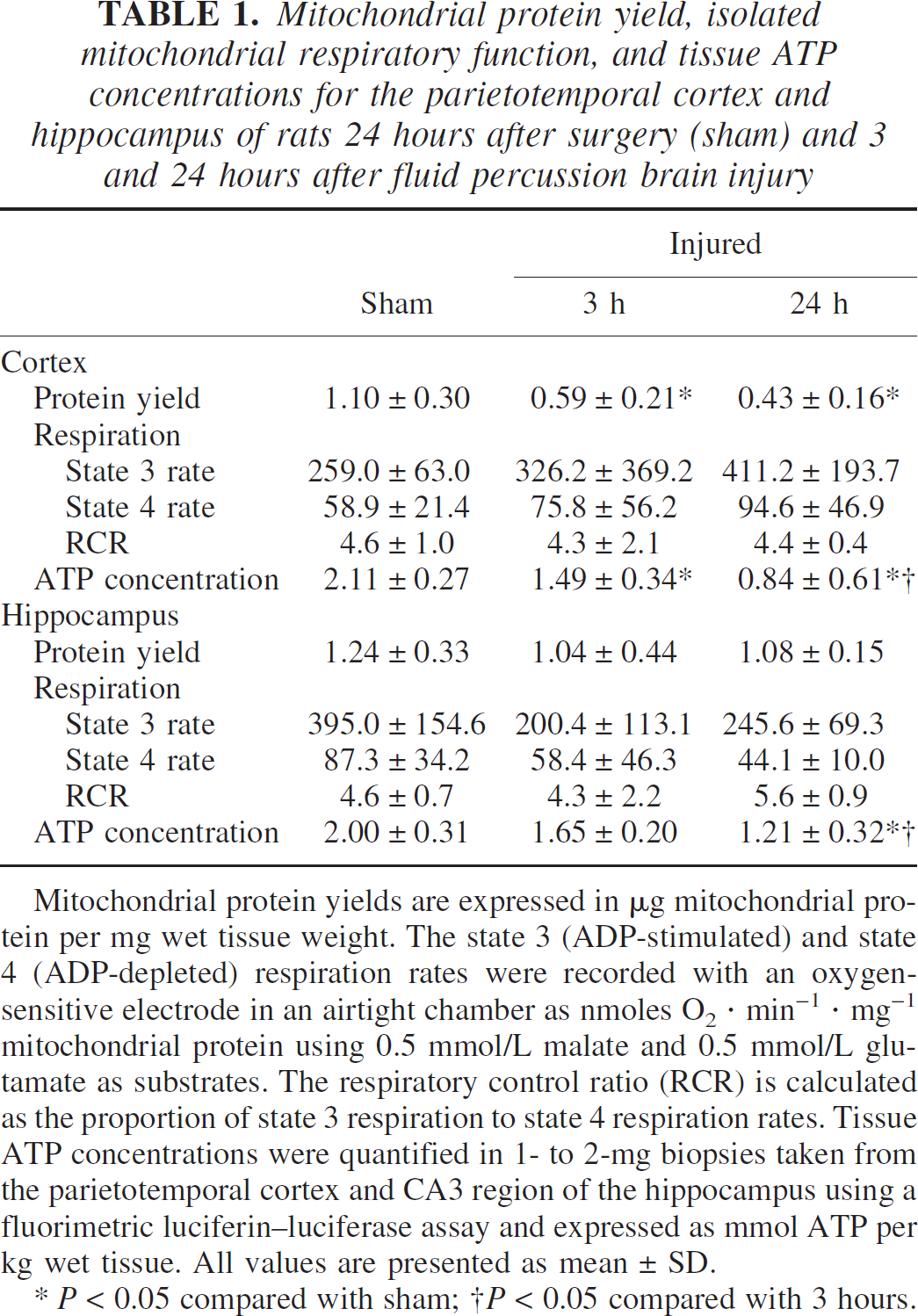
Mitochondrial protein yields are expressed in μg mitochondrial protein per mg wet tissue weight. The state 3 (ADP-stimulated) and state 4 (ADP-depleted) respiration rates were recorded with an oxygen-sensitive electrode in an airtight chamber as nmoles O2 · min−1 · mg−1 mitochondrial protein using 0.5 mmol/L malate and 0.5 mmol/L glutamate as substrates. The respiratory control ratio (RCR) is calculated as the proportion of state 3 respiration to state 4 respiration rates. Tissue ATP concentrations were quantified in 1- to 2-mg biopsies taken from the parietotemporal cortex and CA3 region of the hippocampus using a fluorimetric luciferin—luciferase assay and expressed as mmol ATP per kg wet tissue. All values are presented as mean ± SD.
P < 0.05 compared with sham
P < 0.05 compared with 3 hours.
Respiratory function remains coupled in mitochondria isolated after traumatic brain injury
To assess whether mitochondrial functional deficits were evident after FP brain injury and isolation, we evaluated the rates of oxygen consumption and respiratory coupling to ADP during the phosphorylation of ADP to ATP driven by the complex I substrates malate and glutamate. The oxygen consumption rates during state 3 and state 4 respiration and the respiratory control ratios in mitochondria isolated from the cortex and hippocampus of sham (uninjured) animals were in accordance with previously published values using similar mitochondrial isolation protocols (Table 1) (Sims, 1990; Friberg et al., 1999). No significant differences were detected in state 3 or state 4 respiration rates between mitochondria isolated from injured and control (uninjured) brain regions (cortex or hippocampus), demonstrated further by comparable respiratory control values (Table 1). The state 3 and state 4 rates of oxygen consumption, however, suggest a trend towards increased respiratory activity in the cortical population and respiratory depression in the hippocampal population from injured brain, which did not achieve significance.
Tissue ATP concentrations decline in the cortex and hippocampus after traumatic brain injury
To minimize the potential confounds associated with mitochondrial isolation and experimental conditions, metabolic function was indirectly assessed in vivo by measuring tissue ATP concentrations in tissue biopsies from the selectively vulnerable parietotemporal cortex and CA3 region of the hippocampus after TBI. The ATP concentration in tissue biopsies from the cortex of uninjured (sham) animals was approximately 2 mmol/kg wet tissue weight, in accordance with previously published values (Table 1) (Hylton et al., 1995). After brain injury of moderate severity, ATP concentrations in the parietotemporal cortex at 3 hours were 30% lower than uninjured values (P < 0.05 vs. sham), and decreased further to 60% below uninjured values by 24 hours (P < 0.05 vs. 3-hour injured and sham). In the ipsilateral CA3 region of the hippocampus, ATP concentrations were unchanged at 3 hours, but were decreased by 40% at 24 hours after injury (P < 0.05 vs. 3-hour injured and sham). Tissue ATP concentrations in the CA1 region of the hippocampus were unaffected by brain injury (data not shown), as opposed to the more vulnerable CA3 region. Correcting these values to account for the edema associated with FP brain injury (Soares et al., 1992) does not eliminate the differences between injured and uninjured groups (data not shown).
Greater proportions of mitochondria isolated from injured brain exhibit ultrastructural damage
To assess whether mitochondria are morphologically damaged after brain injury, electron microscopy was performed on isolated mitochondria because mitochondrial morphologic damage indicates associated pathology (Hackenbrock, 1966; Schweiger et al., 1988). Electron micrographs of mitochondrial populations isolated from the parietotemporal cortex and the hippocampus after TBI or sham injury are presented in Fig. 2, in which synaptosomes and membrane debris were infrequently observed. Mitochondria from sham (uninjured) animals (Figs. 2A and 2D) typically exhibit round profiles with discernible inner and outer membranes. In contrast, mitochondria isolated from both injured brain regions showed a variety of mitochondrial sizes, shapes, and internal structures (Figs. 2B, 2C, 2E, and 2F). Morphologically damaged mitochondria had disrupted cristae, thinned outer membranes, and had a ballooned appearance (arrows in Figs. 2B, 2C, 2E, and 2F). Because the release of apoptogenic proteins depends on the rupture of the outer mitochondrial membrane (He and Lemasters, 2002; Brustovetsky et al., 2002), the membrane debris (star in Fig. 2C) and mitochondria that lack long segments of outer membrane (arrowheads in Fig. 2B, 2C) represent injury-associated pathology to the isolated mitochondrial population.
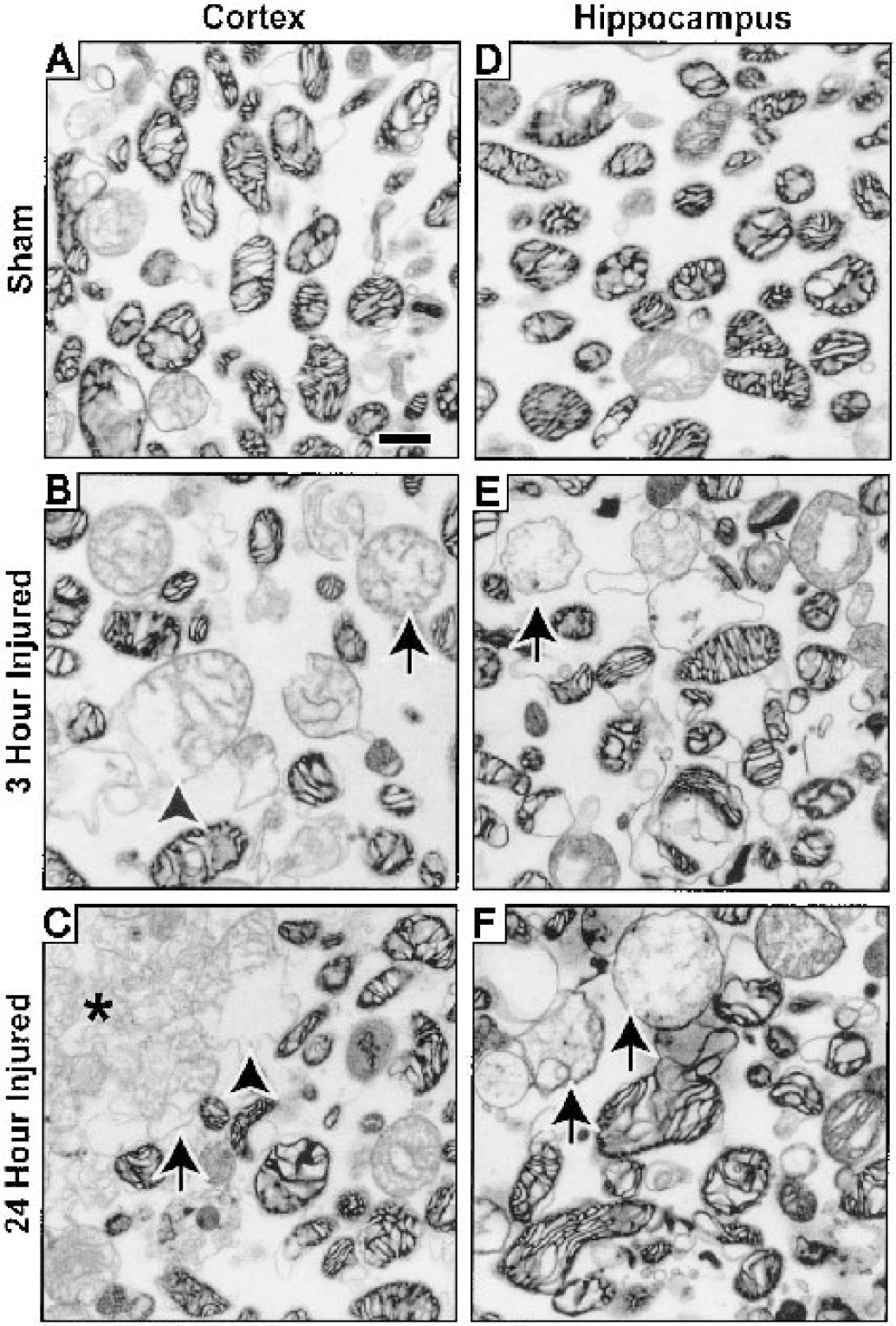
Ultrastructure of mitochondria isolated from the parietotemporal cortex
To express the proportions of the mitochondrial populations that exhibited ultrastructural damage, we categorized mitochondrial morphology as condensed (compacted matrix cristae and continuous outer membrane, regardless of size or shape) or damaged (fragmented cristae with evidence of an expanded matrix compartment or disrupted outer membrane). Mitochondria from the cortex and hippocampus of uninjured animals were found to be predominantly of condensed morphology, showing negligible damage associated with isolation and fixation (Fig. 3A), as previously reported (Brustovetsky et al., 2002). The proportion of damaged mitochondria in the populations isolated from brain-injured animals was significantly increased 3 hours and 24 hours after injury in both the parietotemporal cortex and hippocampus compared with uninjured samples (P < 0.05).
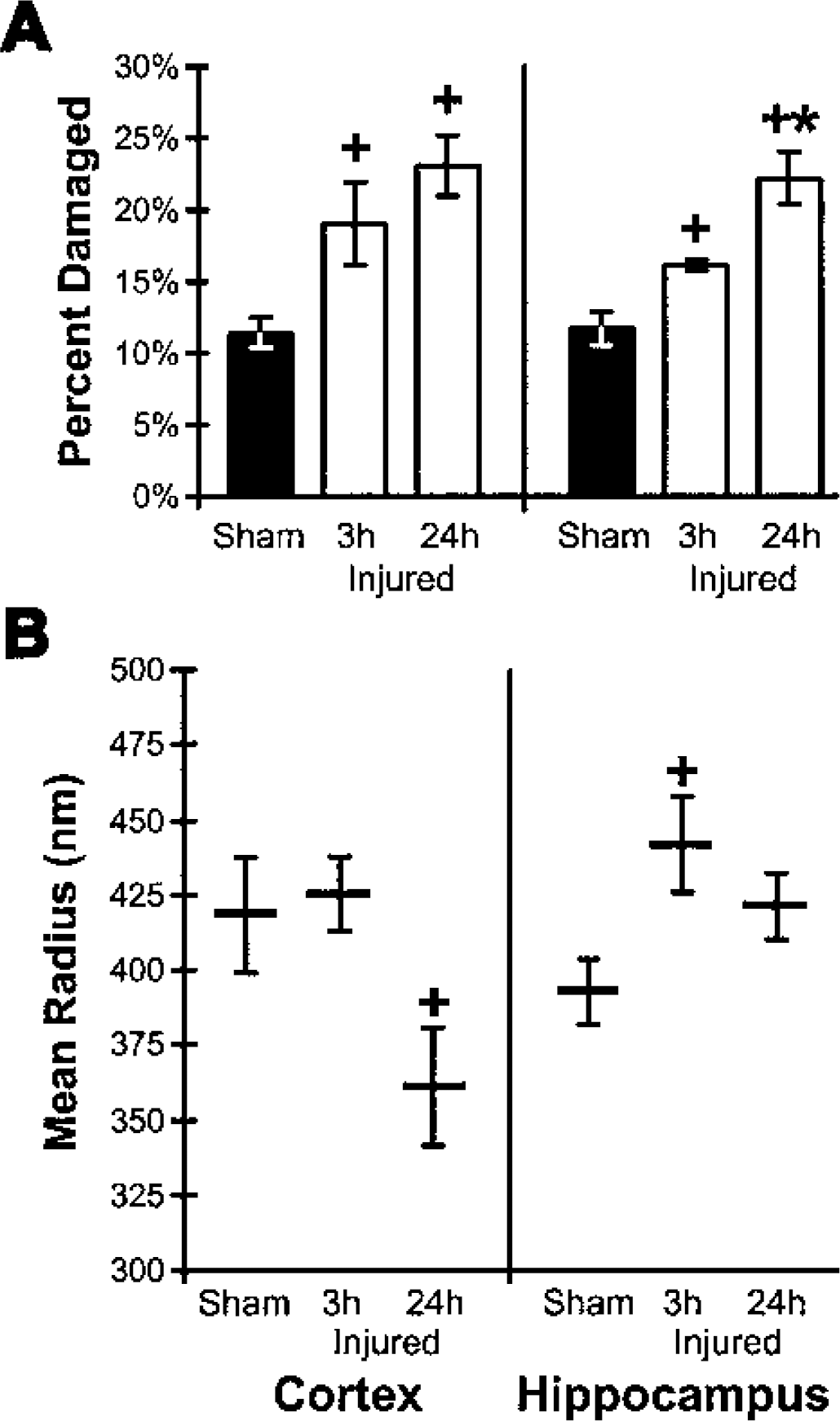
Structural quantification of the mitochondrial populations isolated from the parietotemporal cortex and hippocampus of brain-injured and uninjured (sham) animals as reflected by morphologic damage and mean mitochondrial size.
Smaller cortical and larger hippocampal mean mitochondrial size after traumatic brain injury
Quantitative assessment of mitochondrial size using photon correlation spectroscopy showed that the mean radius of mitochondria in the populations isolated from uninjured or injured brain was in accordance with previously reported values (Hackenbrock, 1966; Kristian et al., 2000). Although unchanged at 3 hours, the mitochondrial population isolated from the parietotemporal cortex 24 hours after injury exhibited a significantly smaller mean radius (13.7%) compared with the population from uninjured animals (P < 0.05, Fig. 3B). In the hippocampus, the mean mitochondrial radius of the population isolated 3 hours after injury was significantly larger (12.4%) than that of uninjured animals (P < 0.05, Fig. 3B). At 24 hours after injury, the mean mitochondrial radius of the hippocampal population remained elevated, but was no longer significantly different from uninjured values.
Calcium-induced swelling rates are slower in mitochondria isolated after traumatic brain injury
To assess the kinetics and mechanics of MPT pore activation, isolated mitochondria were challenged by exposure to increasing concentrations of exogenous calcium under deenergized conditions. Because in vitro respiratory coupling was equivalent between injured and uninjured samples (Table 1), deenergized conditions eliminated compensation of the calcium exposure or free radical generation by respiratory activity (Votyakova and Reynolds, 2001). The rate of calcium-induced MPT pore activation in isolated mitochondria was detected as a decrease in light scatter over time due to osmotic swelling (Fig. 4A). Similar patterns in the response of mitochondrial populations to exogenous calcium were observed in isolates from the parietotemporal cortex (Fig. 4B) and hippocampus (Fig. 4C). Under deenergized conditions, isolated mitochondria exhibited osmotic swelling in the absence of calcium, with no significant increase in swelling rate induced by 20 μmol/L calcium in uninjured or injured samples. Above 20 μmol/L calcium, swelling rates increased in a dose-dependent manner when mitochondria were exposed to calcium concentrations up to 200 μmol/L, representing the increasing probability of the MPT pore opening within the calcium-sensitive population. Mitochondria isolated from the cortex and hippocampus of brain-injured animals also exhibited dose-dependent increases in swelling rates with the addition of calcium above 20 μmol/L (Figs. 4B and 4C). However, the swelling rates of mitochondria from both the cortex and hippocampus of injured animals were significantly slower than those observed in mitochondrial populations from uninjured animals (P < 0.05 at 50, 100, and 200 μmol/L calcium).
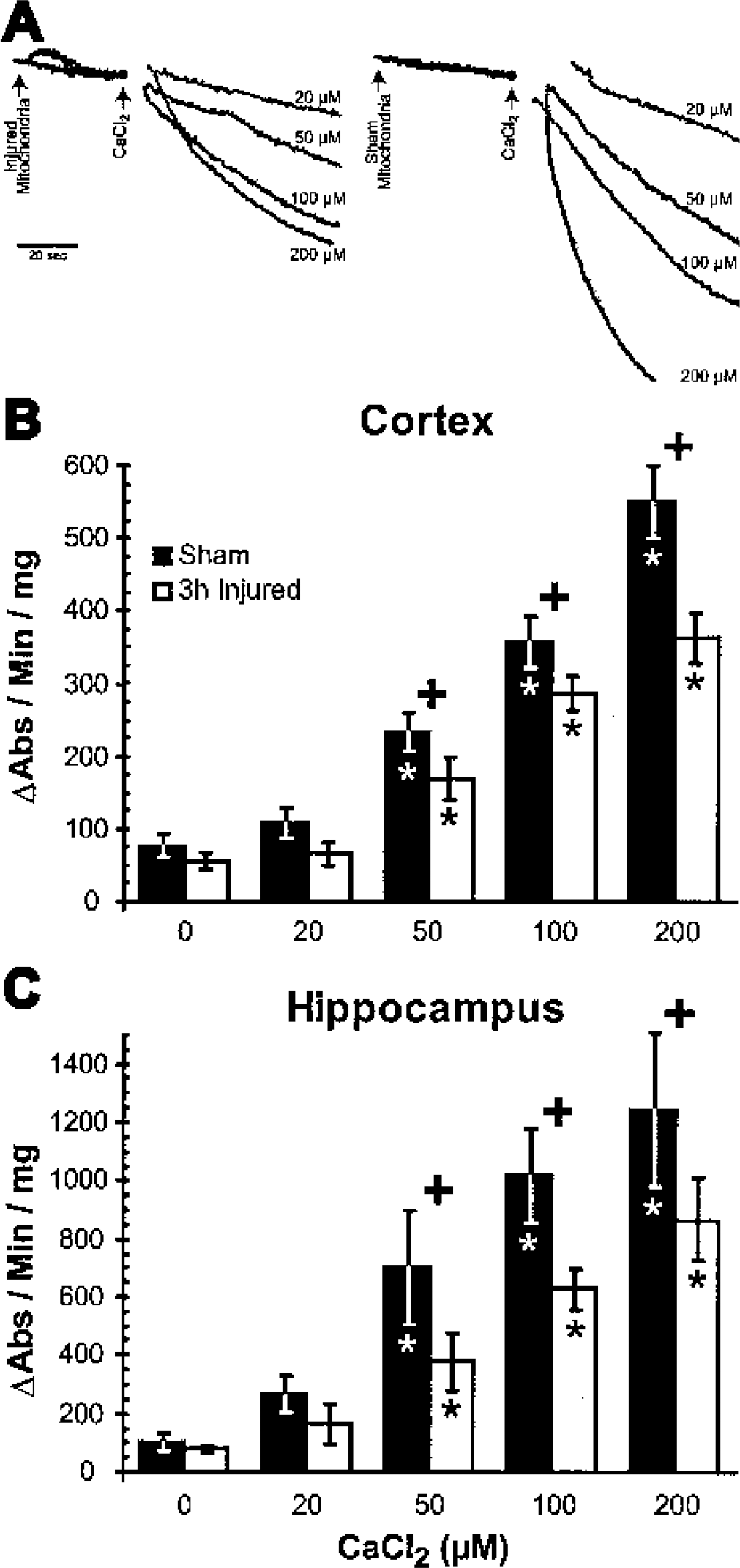
Calcium-induced swelling rates of mitochondria isolated from the parietotemporal cortex and hippocampus of brain-injured and uninjured (sham) rats in response to a calcium challenge under deenergized conditions.
Calcium-induced swelling in mitochondria isolated from injured brain is predominantly mediated through regulated mitochondrial permeability transition pores
To discriminate the contributions of regulated and unregulated MPT pores to the calcium-induced swelling (He and Lemasters, 2002), CsA-insensitive (unregulated, high-conductance MPT pore) swelling rates were measured in the presence of the MPT pore inhibitor CsA (Fig. 5A). The CsA-sensitive (regulated, low-conductance MPT pore) swelling rates were calculated as the difference between total swelling and CsA-insensitive swelling. In parietotemporal cortical mitochondria (Fig. 5B), CsA-sensitive swelling rates at 100 μmol/L calcium were significantly faster than at 50 μmol/L calcium (P < 0.05). For mitochondria isolated from the uninjured parietotemporal cortex and exposed to 50 μmol/L or 100 μmol/L calcium, the CsA-sensitive component of the total calcium-induced swelling rate was significantly greater than the CsA-insensitive component (77 ± 9% for 50 μmol/L, 78 ± 12% for 100 μmol/L, P < 0.05, Fig. 5B), verifying that calcium-induced swelling is mediated primarily through low-conductance, regulated MPT pores. The CsA-insensitive swelling rates were significantly slower in injured than uninjured samples (P < 0.05), increasing the contribution of CsA-sensitive swelling to the total swelling rate to 88 ± 8% at 50 μmol/L calcium (P = 0.09, Mann-Whitney U test) and 91 ± 19% at 100 μmol/L calcium (P = 0.12, Mann-Whitney U test) of the total swelling rate. In hippocampal mitochondria (Fig. 5C), both the CsA-sensitive and CsA-insensitive swelling rates increased significantly when the calcium concentration was increased from 50 μmol/L to 100 μmol/L (P < 0.05) and both components were significantly slower in injured compared with uninjured samples (P < 0.05). Equivalent injury-induced reductions in both components of the total swelling rate resulted in similar relative contributions of CsA-sensitive swelling to the total swelling rate in uninjured (81 ± 9% for 50 μmol/L, 78 ± 11% for 100 μmol/L) and injured (81 ± 15% for 50 μmol/L, 81 ± 6% for 100 μmol/L) hippocampal samples, negating an interaction between the injury and the components of calcium-induced swelling. The contribution of CsA-sensitive swelling to the total calcium-induced swelling rates for both cortical and hippocampal samples from uninjured and brain-injured animals was not dependent on calcium concentrations, suggesting that the distribution of regulated and unregulated MPT pores in CNS mitochondrial populations is unaffected by calcium and may be a defining characteristic of mitochondrial populations isolated from brain.
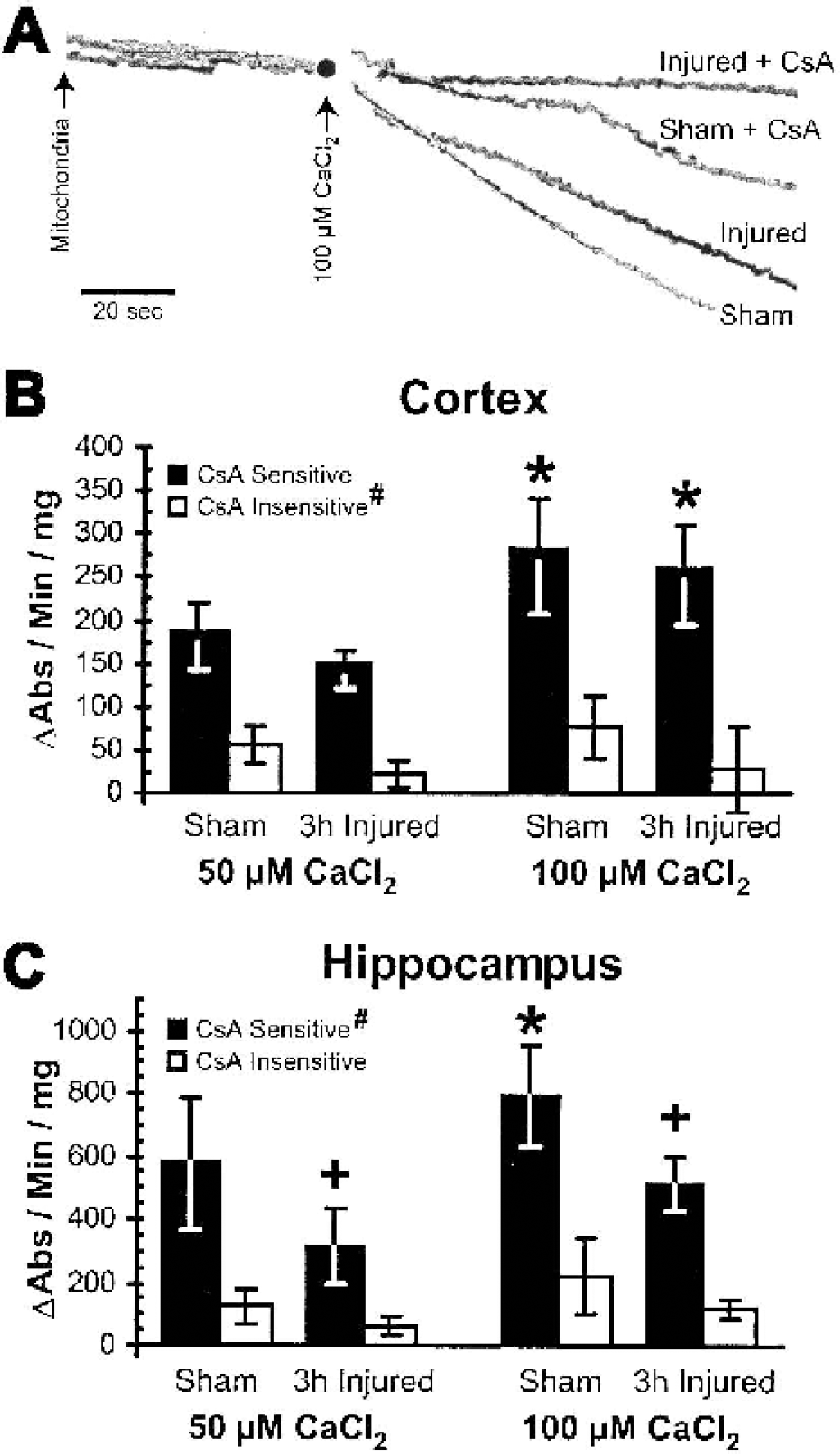
Cyclosporin A (CsA) sensitivity of the total swelling rates in response to 50 μmol/L and 100 μmol/L CaCl2 in mitochondria isolated from the parietotemporal cortex and hippocampus of uninjured (sham) and brain-injured animals.
Population model segregates central nervous system mitochondria by sensitivity to injury, calcium, and cyclosporin A
To interpret the response of mitochondrial populations to TBI, we have developed a model of the total mitochondrial population that segregates isolated mitochondria by morphology, calcium sensitivity, and CsA sensitivity (Fig. 6A). The total mitochondrial populations within the cortex and hippocampus are susceptible to the injury and/or isolation process, such that a portion of the population is eliminated and cannot be assessed in vitro. We believe that the reduction in mitochondrial protein yield from injured tissue (Table 1) identifies the portion of the total population eliminated as a result of injury. Furthermore, the isolated mitochondrial population can be segregated by morphology to reveal the relative proportions of damaged and condensed mitochondria (Figs. 2 and 3A). By incorporating the reduced mitochondrial yield after injury (Table 1) and the ratios of mitochondrial morphologies in the isolated population (Fig. 3A), a conserved percentage (∼10%) of the total mitochondrial population from the parietotemporal cortex exhibits damage, as increasingly more condensed mitochondria are eliminated as a result of injury (Fig. 6B). The morphologic damage in the mitochondrial populations isolated from the hippocampus arises primarily from a reduction in the condensed mitochondrial population (Fig. 6B). The reduction in calcium-induced swelling rates in mitochondria isolated from brain-injured versus uninjured tissue suggests that the total mitochondrial population may be further segregated by the calcium sensitivity of MPT pore activation (calcium gating potential) (Figs. 4 and 6A). The CsA sensitivity of the calcium-sensitive population reflects the proportions of the population that exhibit the regulated (CsA-sensitive) and unregulated (CsA-insensitive) MPT pores (Figs. 5 and 6A).
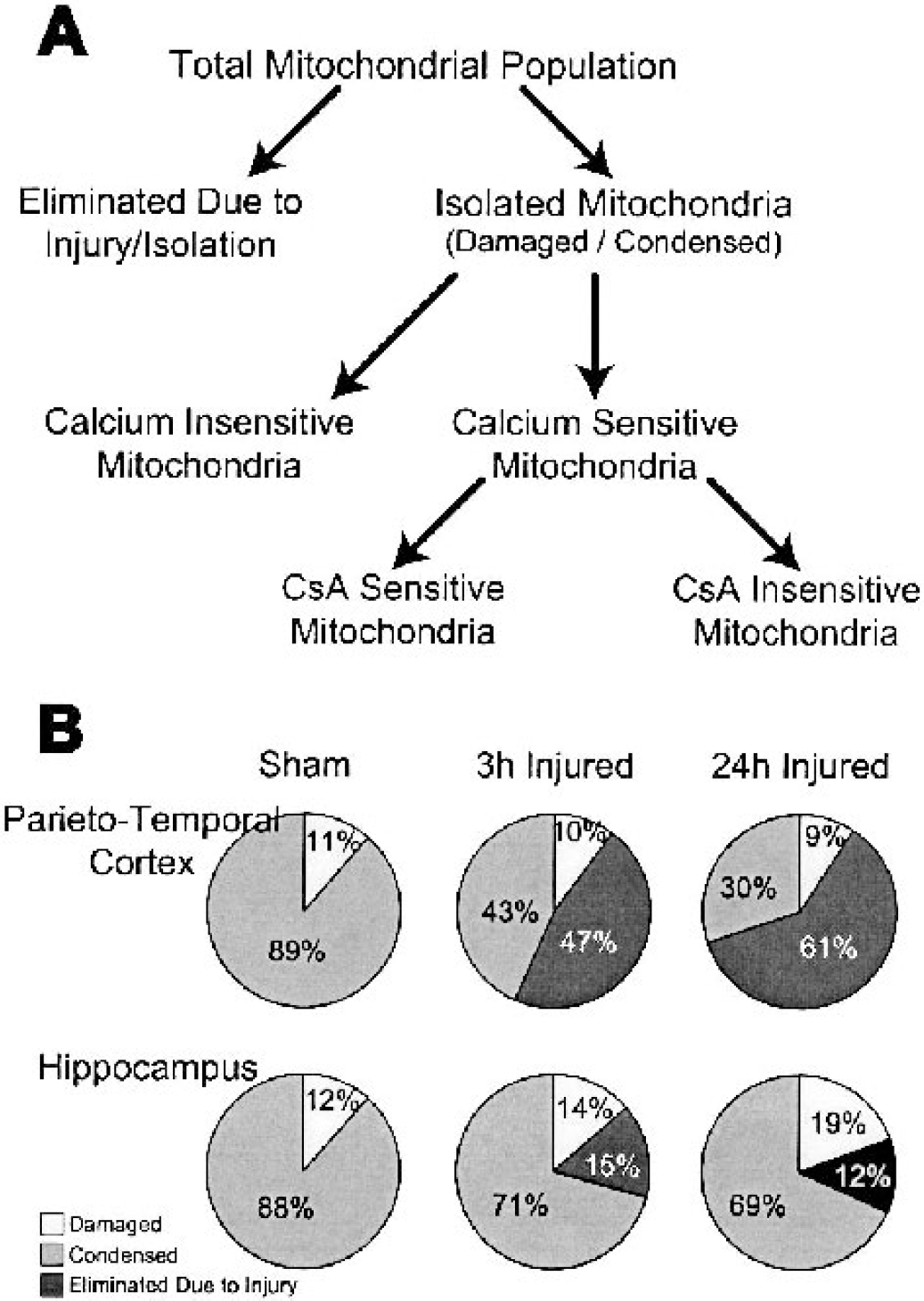
Model of the CNS mitochondrial population segregated by sensitivities to injury, isolation, calcium, and cyclosporin A (CsA).
DISCUSSION
Within 24 hours, experimental TBI alters the structural properties and MPT pore regulation of the mitochondrial populations isolated from the injured parietotemporal cortex and ipsilateral hippocampus as evidenced by reduced mitochondrial protein yield, disruption of membrane structure, shifts in mean radius, altered calcium-induced MPT pore activation, and differential CsA sensitivity. Because technical difficulties limit direct evaluation and manipulation of the mitochondrial population in vivo, we developed a model for the mitochondrial population response to TBI, based on the properties of mitochondria isolated from brain-injured compared with uninjured rat brain. With the reductions in protein yield (Table 1), tissue ATP (Table 1), condensed (healthy) mitochondria (Fig. 3A), and mean radius (Fig. 3B) after injury, we suggest that the mitochondrial population from the traumatized cortex may be damaged and eliminated in vivo. The remaining calcium-sensitive isolated population demonstrated less sensitivity to calcium-induced MPT pore activation (Fig. 4B) through CsA-sensitive low-conductance, regulated MPT pores (Fig. 5B). Conversely, the mitochondrial population from injured hippocampus shows structural damage, a larger mean radius (Fig. 3B), and reductions in CsA-sensitive, CsA-insensitive, and total calcium-induced swelling rates after injury (Figs. 4C and 5B), suggesting that the mitochondrial population in the hippocampus may sustain damage and swell in vivo (which becomes evident in vitro). Mitochondria may exist on a continuum between condensed, damaged, and eliminated (Fig. 6A), such that the cortical population has been pushed further toward damage and elimination than the hippocampal population by 24 hours after injury, reflecting the relative delay in the pathology between these brain regions (Dietrich et al., 1994; Hicks et al., 1996).
In the present study, the variability within each experiment attests to the heterogeneity within CNS mitochondrial populations, previously suggested by the sustained calcium accumulation that follows an initial calcium-induced calcium release, the absence of calcium-induced swelling after the initial exposure, membrane potential dependence of CsA efficacy, and sensitivity towards cytochrome c release (Kristian et al., 2000; Brustovetsky and Dubinsky, 2000b; Maciel et al., 2001; Brustovetsky et al., 2002). Based on MPT pore sensitivity to CsA, mitochondrial populations may consist of a continuum of mitochondria with regulated and unregulated MPT pores (Gogvadze et al., 2001; He and Lemasters, 2002), thus defining the population heterogeneity by the state of the MPT pores in terms of CsA sensitivity, conductance, and permeability (Novgorodov and Gudz, 1996). Additionally, the glial infiltration and reactive gliosis that have been documented by 1 day after this model of brain injury could contribute to the heterogeneity observed (Hill et al., 1996). Similarly, synaptosomal preparations increase the neuronal content of isolated mitochondrial populations and show less variability in membrane potential than somal preparations after experimental brain injury (Sullivan et al., 1999).
The reductions in TTC staining, tissue ATP, and mitochondrial yield observed in the present study may be attributed to both in vivo mitochondrial dysfunction and a combination of mitochondrial and cellular loss after injury. Secondary cascades initiated after TBI, including calcium accumulation (Fineman et al., 1993), caspase activation (Buki et al., 2000), and free radical generation (Lewen and Hillered, 1998), may mediate the elimination of mitochondria (Pettus et al., 1994). In fact, apoptosis can selectively eliminate mitochondria in the presence of caspase inhibitors (Xue et al., 2001), and oxidative stress has been shown to reduce mitochondrial protein yield from the liver in a model of chronic ethanol consumption (Cahill et al., 1997). Damage or elimination of mitochondrial populations after injury may underlie the sustained hypometabolic state reported to be initiated within hours after experimental TBI (Vink et al., 1988; Yoshino et al., 1991; Ginsberg et al., 1997; Moore et al., 2000) that is coupled to increased brain lactate concentration (Nilsson et al., 1990; Kawamata et al., 1995) and reduced cerebral blood flow (Yamakami and McIntosh, 1991; Ginsberg et al., 1997), suggesting that cerebral metabolism may shift towards anaerobic glycolysis. The acute decrease in the [ATP]/[ADP][Pi] ratio that arises from increases in cytosolic ADP (Vink et al., 1988; Headrick et al., 1994) and reductions in tissue ATP concentrations (Signoretti et al., 2001)(Table 1) incorporates both a loss of energy production and an increase in energy demand after moderate, but not mild (Yang et al., 1985), brain injury. Cerebral metabolic alterations and mitochondrial damage may contribute to the progressive cell death observed in the parietotemporal cortex and CA3 region of the hippocampus after FP brain injury (Hicks et al., 1996; Smith et al., 1997) that would be accompanied by additional loss of mitochondria. Alternatively, reduced protein yields may reflect TBI-induced mitochondrial alterations that result in a redistribution of mitochondria to other fractions, because the protocol was developed for uninjured tissue. Future studies to address the potential redistribution of mitochondrial populations should focus on alternative isolation protocols and enzymatic analysis of mitochondrial markers within the homogenates.
Respiratory coupling in isolated mitochondria and homogenized tissue (data not shown) was unchanged after TBI, suggesting that substrate and oxygen availability in vitro may mask the metabolic stress evident in altered ATP concentrations and cerebral metabolism after FP brain injury (Yoshino et al., 1991; Ginsberg et al., 1997; Moore et al., 2000). Vink et al. (1990) found no injury-induced changes in hemispheric cortical respiratory coupling after FP brain injury, although sustained bilateral hemispheric deficits have been reported after CCI brain injury in rats (Xiong et al., 1997). This discrepancy between models may originate from the greater severity of injury induced by CCI than FP in terms of blood—brain barrier disruption and lesion volume (Perri et al., 1997; Hicks et al., 1997; Baskaya et al., 2000). However, in vitro mitochondrial function may be preserved despite morphologic damage to mitochondrial membranes (Vander Heiden et al., 2000; Waterhouse et al., 2001).
Ultrastructural morphology shows that larger proportions of the mitochondrial populations isolated from both parietotemporal cortex and hippocampus are damaged acutely after TBI, similar to reports of mitochondrial damage after cerebral ischemia (Schweiger et al., 1988). The shifts in mean mitochondrial size could be interpreted as an elimination of larger mitochondria from the cortical population and the presence of more swollen mitochondria remaining in the hippocampal population. The differences between the morphology and mean size of isolated mitochondria from injured tissue may be reconciled, in part, by our mitochondrial segregation model that suggests a smaller proportion of condensed mitochondria in the cortex and an increasing proportion of damaged mitochondria in the hippocampus (Fig. 6B).
When challenged by exogenous calcium, isolated mitochondrial populations from both the cortex and the hippocampus of brain-injured and uninjured animals exhibited dose-dependent increases in swelling rates, reflecting proportionally more open MPT pores in the population (Petronilli et al., 1993). Complete opening of all MPT pores within a population was not achieved, however, because swelling rates did not plateau at higher calcium concentrations. For injured samples, the shift in the dose-dependent response of calcium-induced swelling rates compared with uninjured samples suggests that the posttraumatic cellular environment may affect calcium sensitivity of the MPT pore in the mitochondrial population, which may arise from modifications in the components of the MPT pore that reduce the number of open MPT pores or the kinetics of activation (Halestrap et al., 1998). Alternatively, mitochondria isolated from injured brain may (1) preferentially burst when exposed to calcium, thereby reducing swelling rates; or (2) swell more slowly if swelling has occurred in vivo. Furthermore, mitochondria that survive the initial in vivo insult may be protected by an upregulation of antiapoptotic Bcl-2 protein (Clark et al., 1997; Raghupathi et al., 2000), which can translocate to mitochondria and increase calcium-buffering capacity or reduce calcium sensitivity (Murphy et al., 1996). Preliminary observations remain inconclusive regarding levels of Bcl-2 protein in mitochondria after TBI (data not shown).
For isolated cortical mitochondria, reductions in total and CsA-insensitive calcium-induced swelling, without a change in CsA-sensitive swelling, suggest that mitochondria with unregulated MPT pores may be those eliminated by injury. For the hippocampus, the reduction in total swelling rate stems from slower CsA-sensitive and CsA-insensitive swelling, indicative of damage throughout the population. Thus, in vivo TBI may not drive MPT pores into an unregulated, high-conductance, pathologic state. The weak antagonism of the MPT pore by CsA, however, can be overcome with excessive calcium (Brustovetsky and Dubinsky, 2000b), which was evident for CsA-sensitive swelling. The efficacy of CsA administration to brain-injured animals in protecting isolated mitochondria and CNS tissue, however, attests to the preserved mitochondrial CsA sensitivity after brain injury (Okonkwo and Povlishock, 1999; Sullivan et al., 1999; Riess et al., 2001).
The heterogeneity within CNS mitochondrial populations, encompassing varying sensitivities to calcium and CsA, may underlie the regional cell death associated with TBI. Selective elimination of mitochondria due to injury or isolation may arise from extensive oxidative membrane damage (Lewen and Hillered, 1998; Tyurin et al., 2000), in vivo swelling similar to cytotoxic edema (Soares et al., 1992), or the primary impact. Differential calcium sensitivity may arise from direct (free radical oxidation, proteolysis) or indirect (steric hindrance, acidosis) damage to the putative calcium sensor of the MPT pore. Alternatively, cellular responses associated with experimental TBI, such as Bcl-2 translocation (Clark et al., 1997; Raghupathi et al., 2000) or matrix calcium phosphate formation (Wuthier et al., 1985), may influence mitochondrial calcium buffering capacity (Murphy et al., 1996). Similarly, CsA sensitivity may be attributed to differential penetration of CsA into the mitochondrial matrix or the affinity of cyclophilin D for CsA (Tanveer et al., 1996). Determining the specific mitochondrial properties that contribute to TBI pathology necessitates either more specific isolation protocols or techniques capable of evaluating individual mitochondria rather than populations.
Whether damage to and elimination of the mitochondrial populations arises from MPT pore activation in vivo remains to be determined. One cannot say with certainty whether these structural or functional differences occurred in vivo as a result of TBI, or whether the observed alterations occurred as a result of the mitochondrial preparation, which results in a more sensitive population isolated from the injured brain. The injury-induced reduction in calcium sensitivity for both cortical and hippocampal mitochondrial populations, however, provides evidence of mitochondrial pathology that may contribute to the cell death associated with TBI. Through a further understanding of the differential susceptibilities and sensitivities of mitochondria in brain regions vulnerable to TBI, targeted pharmacology may preserve both the mitochondria and CNS tissue.
Footnotes
Acknowledgments
The authors thank Ms. Neelima Shah, for technical support in electron microscopy (Biomedical Imaging Core Laboratory, Department of Pathology and Laboratory Medicine, University of Pennsylvania); and Drs. John T. Povlishock and David A. Hovda, for insightful discussions.