
Abstract
The mechanism by which brief episodes of cerebral ischemia confer protection (tolerance) against subsequent prolonged ischemic challenges remains unclear, but may involve upregulation of cell injury repair capability. The mitochondrion is a key site for the regulation of cell death pathways, and damage to mitochondrial genes has been linked to a number of neurologic diseases and aging. Therefore, the authors examined the response of the DNA base excision repair (BER) pathway in rat brain mitochondria after either brief (tolerance-inducing) or prolonged (injury-producing) focal cerebral ischemia. Brief (30-minute) middle cerebral artery occlusion (MCAO) induced mild oxidative mitochondrial DNA damage and initiated a prolonged (up to 72-hour) activation above control levels of the principal enzymes of the mitochondrial BER pathway, including uracil DNA glycosylase, apurinic/apyrimidinic (AP) endonuclease, DNA polymerase-γ, and DNA ligase. In contrast, prolonged (100-minute MCAO) ischemia induced more substantial mitochondrial oxidative DNA damage whereas elevation of BER activity was transient (∼1 hour), declining to less than control levels over the course of 4 to 72 hours. These data reveal the differences in BER capacity after brief or prolonged ischemia, which may contribute to the neuron's ability to resist subsequent ischemic insults.
Mitochondrial DNA (mtDNA), an ∼16.5-kb circular genome, encodes the 13 essential proteins of the respiratory oxidative phosphorylation pathway, and contains 2 ribosomal RNA and 22 transfer RNA genes (Wallace et al., 1988). Cellular oxidative stress such as that produced by hydroxyl radicals, nitric oxide, and peroxynitrite can lead to the modification of nucleotide bases (Aruoma et al., 1989; Cadet et al., 2000; Wink et al., 1991). For example, guanosine conversion to 8-hydroxydeoxyguanosine (8-oxodG) (Ames, 1989), the deamination of cytosine to uracil, as well as GC to TA transversion all lead to the incorporation of errors in DNA replication (Cheng et al., 1992; Croteau and Bohr, 1997; Krokan et al., 2000). For several reasons, mtDNA is particularly prone to oxidative base damage. Principally, mitochondria are the largest subcellular source for the production of reactive oxygen species (ROS) via the respiratory chain, consuming about 90% of the cell's oxygen intake, of which about 2% is converted to superoxide and hydrogen peroxide (Richter, 1992). Second, mtDNA is usually distributed in several clusters located in close proximity to the inner mitochondrial membrane where ROS are generated as byproducts of the electron transport chain (Richter et al., 1988; Zastawny et al., 1998); compounding this, mtDNA lacks the protection afforded by histones, which are present within nuclear DNA.
Oxidative damage to mtDNA and its consequences (e.g., point mutation, deletion, insertion, and depletion) may account for many mitochondria-associated diseases, including Alzheimer (Hutchin and Cortopassi, 1995) and Parkinson (Bandmann et al., 1997) diseases, as well as the aging process (Ozawa, 1997; Raha and Robinson, 2000). If damage to mtDNA is sufficient, an apoptotic and/or necrotic cell death program may be initiated in the affected cell (Green and Reed, 1998; Papa and Skulachev, 1997). Little is known, however, concerning the mitochondrion's enzymatic machinery for DNA repair.
Recent studies demonstrate that mitochondria can repair some forms of DNA damage including base damage, single-strand breaks, and apurinic and apyrimidinic (AP) sites (Chen et al., 2000; Driggers et al., 1997; Taffe et al., 1996). The DNA base excision repair (BER) pathway is considered the predominant pathway for repair of oxidative damage to mtDNA and after damage to single bases, such as 8-oxodG and deaminated cytosine (uracil) (Croteau et al., 1997; Croteau and Bohr, 1997). These damaged DNA bases are recognized by DNA glycosylases that cleave the N-glycosylic bond to remove the damaged base, leaving a 3' deoxyribose moiety that is removed by an AP endonuclease, generating a 3' hydroxyl group, which is extended by a DNA polymerase before DNA ligase rejoins the free DNA strands (Bogenhagen, 1999; Bogenhagen et al., 2001; Croteau and Bohr, 1997; Pinz and Bogenhagen, 1998; Seeberg et al., 1995) (see Fig. 1 for an overview). The presence of the enzymes of the BER pathway in rat brain has recently been confirmed (Chen et al., 2000, 2002).
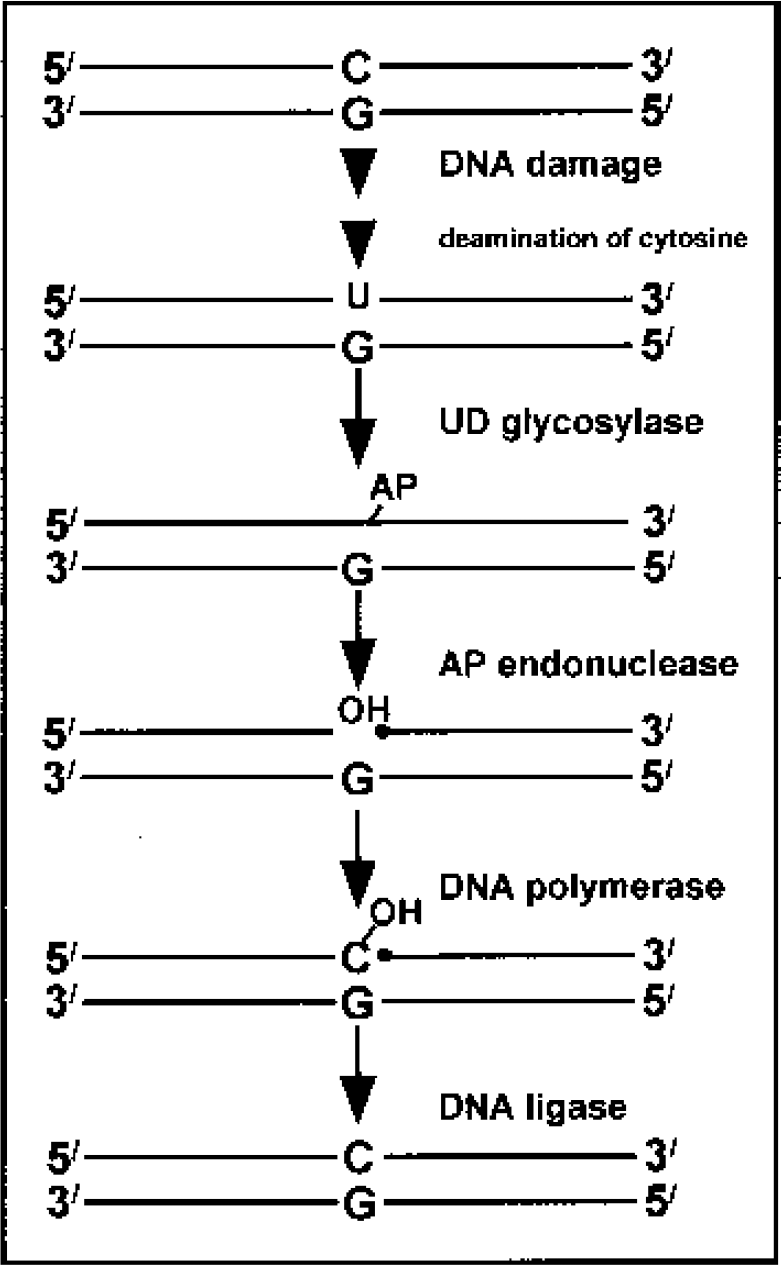
Overview of the base excision repair pathway. Following deamination of cytosine to uracil, the uracil is cleaved by uracil DNA glycosylase to form a free 3' deoxyribose moiety (AP site), which is then removed by apurinic/apyrimidinic (AP) endonuclease. DNA polymerase adds the correct base back to the DNA, and the DNA strands are joined by DNA ligase.
DNA damage is a well-documented consequence of ischemic brain injury (Love, 1999), comprising lesions such as 8-oxodG and single-strand breaks (Chen et al., 1997; Liu et al., 1996; Nagayama et al., 2000b). Further, such DNA damage, particularly when left unrepaired, may trigger cell death pathways or render cells susceptible to subsequent injury (Martin, 2001; Zhou and Elledge, 2000). Brief periods of focal or global cerebral ischemia have been shown to confer protective effects (tolerance) against subsequent prolonged ischemic episodes (Chen and Simon, 1997), but the underlying mechanisms remain elusive. The present study examined the upregulation and engagement of DNA repair mechanisms in mitochondria as a possible mechanism of the underlying protective effects of brief, preconditioning focal cerebral ischemia.
MATERIALS AND METHODS
Reagents
HEPES, ATP, bovine serum albumin, β-mercaptoethanol, phosphocreatine, creatine phosphokinase, DTT, SDS, NAD+, and unlabeled deoxynucleoside triphosphates (dNTPS) were purchased from Sigma (St. Louis, MO, U.S.A.). Radiolabeled NTPs and dNTPs were obtained from NEN (Boston, MA, U.S.A.); Klenow polymerase, T4 DNA ligase, and uracil DNA glycosylase (UDG) were purchased from Gibco BRL (Rockville, MD, U.S.A.); and G-25 spin columns were obtained from 5 prime-3 prime Inc. (Boulder, CO, U.S.A.).
Oligonucleotides
The following oligonucleotides were synthesized by Bio-Synthesis: M50U (5'-TCGGTACCCGGGGATCCTCTAGAGTUGACCTGCAGGCATGCAAGCTTGGC-3'; 3'-AGCCATGGGCCCCTAGGAGATCTCAGCTGGACGTCCGTACGTTCGAACCG-5'); M58U (self-annealed oligonucleotide; 5'-TCAATAGTCGACAAGUTTGTGGATCCCCA; 3'-AGTTATCAGCTGTTCGAACACCTAGGGGT); M26P (5'-TCGGTACCCGGGGATCCTCTAGAGTC-3'; 3'-AGCCATGGGCCCCTAGGAGATCTCAGCTG GACGTCCGTACGTTCGAACCG-5'); and T18 (5'-TTTTTTTTTTTTTTTTTTT-3').
Oligonucleotides containing a U site, polyT18, and M26P were labeled using T4 polynucleotide kinase and γ[32P]ATP, purified using a G-25 spin column and stored at a final concentration of 10 μmol/L. Complementary oligonucleotides were annealed in 100 mmol/L KCl (10 mmol/L), Tris pH 7.8, and 1 mmol/L ethylene diamine tetraacetic acid (EDTA) by heating the oligonucleotides to 92°C for 5 minutes, incubating for 15 minutes at 55°C to 70°C, and cooling to room temperature.
Focal cerebral ischemia model of ischemic tolerance or ischemic injury
Middle cerebral artery (MCA) occlusion was induced in adult male Sprague-Dawley rats (280 to 350 g) using the intraluminal filament technique as previously described (Chen et al., 1996). Rats were anesthetized with isoflurane and ventilated to maintain blood gas variables within physiologic range, and rectal and temporalis muscle temperatures were kept at 37.0°C to 37.5°C. A 3–0 surgical monofilament nylon suture with a rounded tip was introduced into the left internal carotid artery and advanced 20 or 21 mm past the carotid bifurcation to occlude the MCA for 30 (tolerance) or 100 minutes (injury inducing) and then withdrawn to allow reperfusion. Animals were killed 15 minutes or 1, 4, 24, or 72 hours after reperfusion or after 4 hours (nonischemic controls).
Isolation of mitochondria and mitochondrial proteins
Mitochondria were isolated as previously described (Chen et al., 2000). Briefly, 100 mg tissue was minced and homogenized in 1x mannitol-sucrose-HEPES-EDTA/EGTA (M-SHE) buffer containing 1 mmol/L dithiothreitol (DTT), 1 μg/mL leupeptin, 1 μg/mL aprotinin, 1 μ/mL pepstatin A, and 1 mmol/L phenylmethylsulfonyl fluoride (PMSF). After lysis for 30 minutes at 4°C, unbroken cells and nuclei were pelleted at 1,200g. The supernatant containing the mitochondria was centrifuged at 10,000g for 15 minutes, resuspended in 3% Ficoll solution, and gently layered over 6% Ficoll solution to produce a discontinuous density gradient. After centrifugation at 10,400g for 25 minutes at 4°C, the sediment was resuspended in 1x M-SHE buffer containing 3 mg/mL digitonin at 4°C for 15 minutes, followed by centrifugation at 10,500g for 15 minutes. The mitochondrial pellet was washed and then lysed for 30 minutes at 4°C by lysis buffer (20 mmol/L HEPES [pH 7.4], 400 mmol/L KCl, 1 mmol/L EDTA, 5% glycerol, 0.5% Triton-X 100, 2 mmol/L DTT, 0.5 mmol/L PMSF, 1 μg/mL aprotinin, 1 μg/mL pepstatin A, and 1 μg/mL leupeptin), centrifuged at 16,000g at 4°C for 30 minutes, concentrated to 5 to 10 mg/mL, and stored at −80°C. This procedure produces a mitochondrial sample free of cytosolic or nuclear proteins (Chen et al., 2000). Fraction purity was verified by Western blotting for the presence of poly (ADP-ribose) polymerase (PARP) as a control for nuclear contamination.
Isolation of mitochondrial DNA and genomic DNA
Purified mtDNA and genomic DNA were isolated according to the manufacturer's instructions (QIAamp DNA Mini Kit). Briefly, 25 mg brain tissue was homogenized and incubated with buffer ATL, proteinase K, and RNase A at 56°C for 3 hours. The sample was diluted, incubated at 70°C for 10 minutes, precipitated with ethanol, and transferred to a QIAamp spin column. The column was centrifuged at 6,000g for 1 minute and then washed. Finally, DNA was eluted from the column with 400 μL TE buffer and the DNA concentration determined spectrophotometrically at A260.
Detection of mitochondrial DNA damage
For detection of mtDNA damage, a quantitative polymerase chain reaction (QPCR) assay was carried out as described previously (Wang et al., 2000) with some minor modifications. The primer for amplification of the rat mtDNA (sense and antisense) was based on the rat mtDNA sequence (Gene Bank Nc001665). Briefly, PCR was initiated in a hot-start at 72°C using the expanded 20-κB plus PCR system (Boehringer Mannheim, Indianapolis, IN, U.S.A.). Twenty-five nanograms total DNA was added in a 50-μL PCR reaction system with 25 cycles (10 seconds at 94°C, 12 minutes at 72°C) and 1 μCi 3,000 Ci/mmol/L [α-32P]dCTP, 200 umol/L dNTP, 20 pmol primer, and 2.5 U Taq enzyme. Amplification products were detected by running on 4% polyacrylamide gels.
Preparation of DNA repair substrates
The BER repair assay was performed using pcDNA3.1 containing the oxidative adduct 8-oxodG, which was originally described by Wood et al. (1989) and later modified by Chen et al. (2000). Briefly, plasmids were grown in E. coli DH5α cells (Gibco) and purified using a large-scale DNA purification Kit (Promega, Madison, WI, U.S.A.) according to the manufacturer's instructions. The closed circular fraction of pCDNA3.1 plasmids was isolated via CsCl gradient centrifugation. CsCl and ethidium bromide were removed by subsequent 1-butanol saturation and extraction with water followed by ethanol precipitation. The DNA was resuspended in TE (pH 8.0) and stored at −20°C. For the induction of 8-oxodG in the plasmid DNA, we used photoactivated methylene blue (MB) as the lesion inducer (Floyd and Carney, 1992). The purified pcDNA3.1 plasmids were diluted to 100 μg/mL, and 1/9 volume 20 μmol/L MB was added to the DNA solution in the dark. The uncovered dish was exposed to light from a 100-W tungsten light bulb for 30 minutes. The DNA was ethanol-precipitated three times to remove MB and then resuspended in TE buffer and stored at −80°C.
In vitro DNA repair assay
In vitro repair assays were performed as previously described (Chen et al., 2000, 2002). Briefly, crude mitochondrial extracts were incubated with 0.3 μg damaged pcDNA3.1 plasmids in a buffer containing 20 μmol/L each of dATP, dTTP, and dCTP, 8 μmol/L dGTP, 2 μCi [32P] dGTP, 40 mmol/L phosphocreatine, and 25 μg creatine phosphokinase at 32°C. The reaction was terminated by the addition of proteinase K (240 μg/mL), 1% SDS, and 20 mmol/L EDTA for 30 minutes at 37°C. Plasmid DNA was purified using Wizard plasmid purification kit, treated with 10 U BamHI at 37°C for 2 hours and separated by electrophoresis on a 1% agarose gel. DNA bands on the gel were visualized using ultraviolet light. Autoradiogram signals on the films were collected using an Optronics DEI-750 3-chip camera equipped with a BQ 8000 sVGA frame grabber and semiquantified (optical density x area) using Bioquant (Nashville, TN, U.S.A.). All densitometric values for DNA radiolabels were normalized to values for ultraviolet photographs of the DNA band on the same lane (Fig 3).
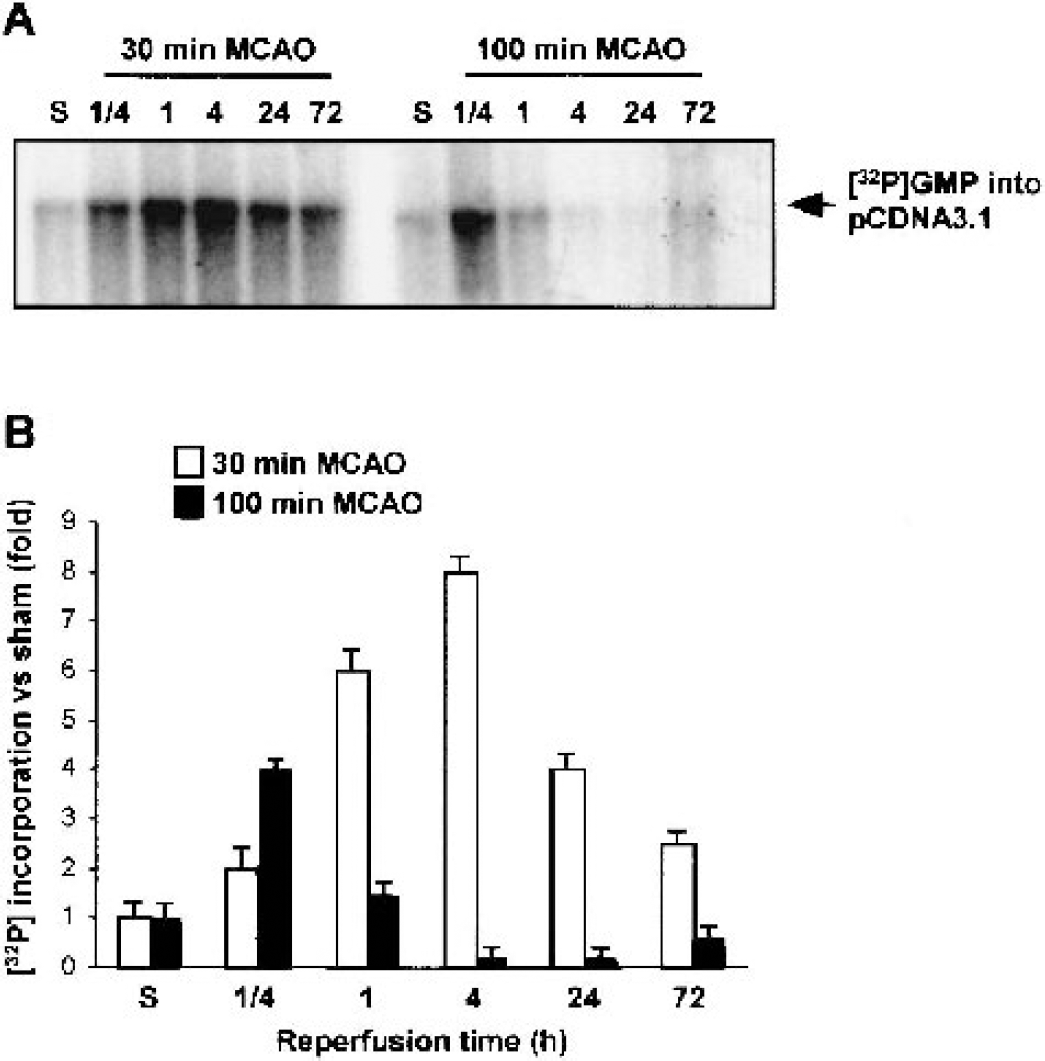
Detection of mitochondrial DNA base excision repair ability of 8-oxodG adduct.
To assess BER repair of uracil bases, mitochondrial extracts (20 μg) were incubated with M50U oligonucleotide in 45 mmol/L HEPES-KOH (pH 7.8), 70 mmol/L KCl, 7.4 mmol/L MgCl2, 0.9 mmol/L DTT, 0.4 mmol/L EDTA, 2 mmol/L ATP, 20 mmol/L each of dATP, dTTP, and dGTP, 8 μmol/L dCTP, 2 μCi [32P] dCTP, 40 mmol/L phosphocreatine, 25 μg creatine phosphokinase, 20 μg/mL bovine serum albumin (BSA), 3.4% glycerol, and 2 mmol/L NAD+. The reaction products were separated by electrophoresis in a 15% polyacrylamide gel containing 7 mol/L urea. After electrophoresis at 200 V for 3 hours, bands were detected by autoradiography using Kodak X-OMAT AR film.
Uracil DNA glycosylase activity assay
Mitochondrial uracil DNA glycosylase (mtUDG) activity was measured by incubating 20 μg extract with 2 pmol of the end-labeled, self-annealed M58U oligonucleotide that was previously digested with BamHI (Fig. 4A). The reaction was carried out in a 20 μL reaction volume in buffer solution of 50 mmol/L HEPES-KOH (pH 7.5), 50 mmol/L KCl, 0.1 mg/mL BSA, and 0.05% Triton X-100 for 50 minutes at 32°C. Reactions were terminated by heating at 80°C for 10 minutes in a loading buffer (90% [vol/vol] formamide, 0.1% [weight/vol] bromophenol blue, and 20 mmol/L EDTA). Samples were loaded on 15% polyacrylamide gel containing 7 mol/L urea and electrophoresed at 200 V. Gels were dried and exposed to Kodak X-OMAT AR film.
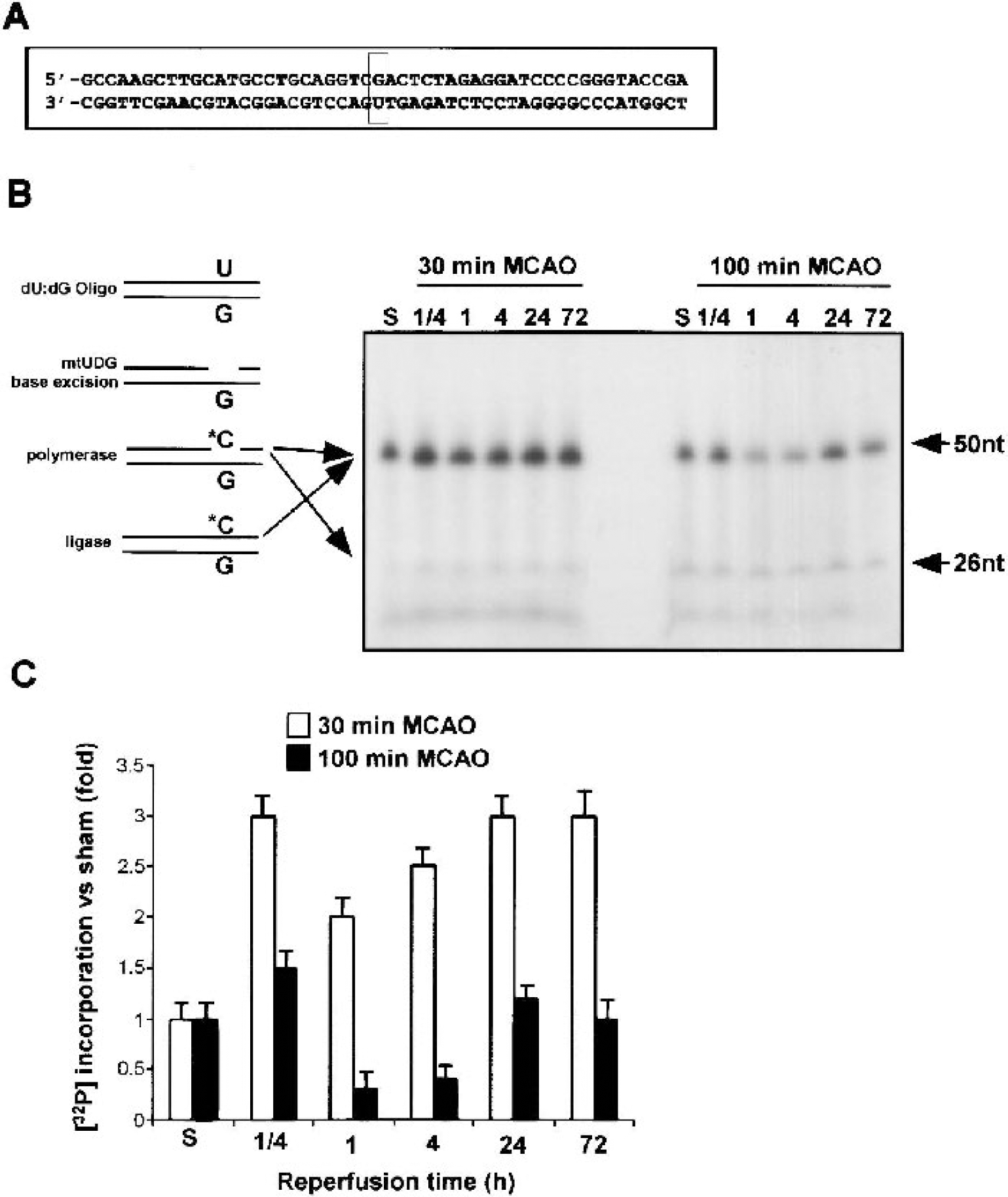
Detection of the ability of mitochondrial DNA base excision repair to replace uracil DNA damage.
Apurinic/apyrimidinic endonuclease assay
An AP site in the M58U oligonucleotide was produced by adding 10 U UDG to 100 pmol M58U self-annealed oligonucleotides at 37°C for 15 minutes. Heating at 55°C for 10 minutes inactivated the UDG. To assay mitochondrial AP endonuclease activity, 10 μg mitochondria extract was incubated with 2 pmol AP-oligonucleotide assay in 20 μL buffer containing 20 mmol/L Tris (pH 8.0), 20 mmol/L KCl, 2 mmol/L CaCl2, 2 mmol/L DTT, 20 μmol/L Zn2+ acetate, 10% glycerol, 0.05% Triton X-100, and 10 μg/mL BSA. Reaction mixtures were incubated for 30 minutes at 32°C and terminated by heating at 55°C for 10 minutes in a loading buffer containing 90% (vol/vol) formamide, 0.1% (weight/vol) bromophenol blue, and 20 mmol/L EDTA. Samples were run on 15% polyacrylamide gel containing 7 mol/L urea and detected by autoradiography.
Mitochondrial DNA polymerase assay
Measuring the extension of the M26P double-stranded oligonucleotides assessed the activity of mitochondrial DNA polymerase. Mitochondrial extracts (20 μg) were incubated with the M26P probe (100 fmol) in a total volume of 20 μL containing 45 mmol/L HEPES-KOH (pH 7.8), 70 mmol/L KCl, 4 mmol/L MgCl2, 1 mmol/L DTT, 1 mmol/L EDTA, 200 μg/mL BSA, and 20 μmol/L each of dATP, dGTP, and dCTP. The reaction was continued at 32°C for 50 minutes, heated to 80°C, and run on a 15% polyacrylamide gel containing 7 mol/L urea. The gel was dried and exposed to Kodak film.
Mitochondrial DNA ligase assay
This assay used annealed T18/PolyA as reaction substrate based on previously described methods (Chen et al., 2000). Briefly, 10 μg of mitochondrial extract was incubated with 2 pmol 32P-labeled T18 oligonucleotide (specific activity, 5 × 106 cpm/pmol) and polyA. The ligation reaction was performed at 32°C for 50 minutes and terminated by adding an equal volume of loading buffer containing 90% (vol/vol) form-amide, 0.1% (weight/vol) bromophenol blue, and 20 mmol/L EDTA followed by heating to 80°C for 10 minutes. The reaction products were separated by electrophoresis on a 15% polyacrylamide gel containing 7 mol/L urea. After electrophoresis at 200 V for 3 hours, bands were detected by autoradiography using Kodak film.
Data analysis
All quantitative data are presented as mean ± SD. Volumes of ischemic damage were compared using ANOVA and post hoc Fisher PLSD tests. A level of P < 0.05 was considered statistically significant.
RESULTS
Mitochondrial DNA damage after brief and prolonged ischemia
To characterize the ischemic injury models, infarct volumes were compared between animals subjected to the following ischemic challenges: prolonged, 100-minute MCAO (group 1); brief, 30-minute MCAO (group 2); and 30-minute ischemia followed by 100-minute MCAO after 72 hours (group 3). Quantification of infarct size 24 hours after the last period of ischemia determined that group 1 (n = 4) exhibited a large (286 ± 48 mm3) infarct within cortex ipsilateral to the occluded vessel. In contrast, group 2 (n = 4) exhibited a smaller infarct (125 ± 46 mm3; P < 0.01). Group 3 (n = 4) exhibited ischemic infarcts >50% smaller than that produced by 100-minute MCAO (134 ± 54 mm3; P < 0.01), whereas ischemic damage did not differ (P = 0.81) from that seen in group 2.
The underlying mechanism of such ischemic tolerance paradigms remains unclear, but may involve alterations in the ability of affected neurons to repair damage to mtDNA. To test such a hypothesis, we first examined the effect of ischemia on mtDNA damage using the widely used QPCR method, which has previously been shown to be a sensitive method to assess the extent of DNA damage, repair, or replication in a semiquantitative fashion (Chen et al., 2001; Englander et al., 1999; Ferre, 1992; Jin et al., 2001; Sawyer et al., 2001; Wang et al., 2000). Damage to mtDNA inhibits the PCR amplification of a fragment that covers 96% of the ∼16-kb mitochondrial genome. Initial experiments showed that there was an increase in 16-kb mtDNA amplification when 2 to 50 ng mtDNA was used as the template (Figs. 2A and 2B). Routine QPCR assays use 25 ng mtDNA. After 30-minute MCAO, QPCR amplification of the fragment had decreased by ∼30% at 4 hours, but recovered to 95% of control by 72 hours (Fig. 2C). In contrast, mtDNA repair function as determined by QPCR amplification decreased to 60% of control by 4 hours after 100-minute MCAO and did not recover (Fig. 2C). These results confirm previous reports that ischemia-reperfusion causes mtDNA damage but that reduced mtDNA content was nearly restored to nonischemic levels 24 hours after a 30-minute (but not a 90-minute) ischemic period (Chen et al., 2001).
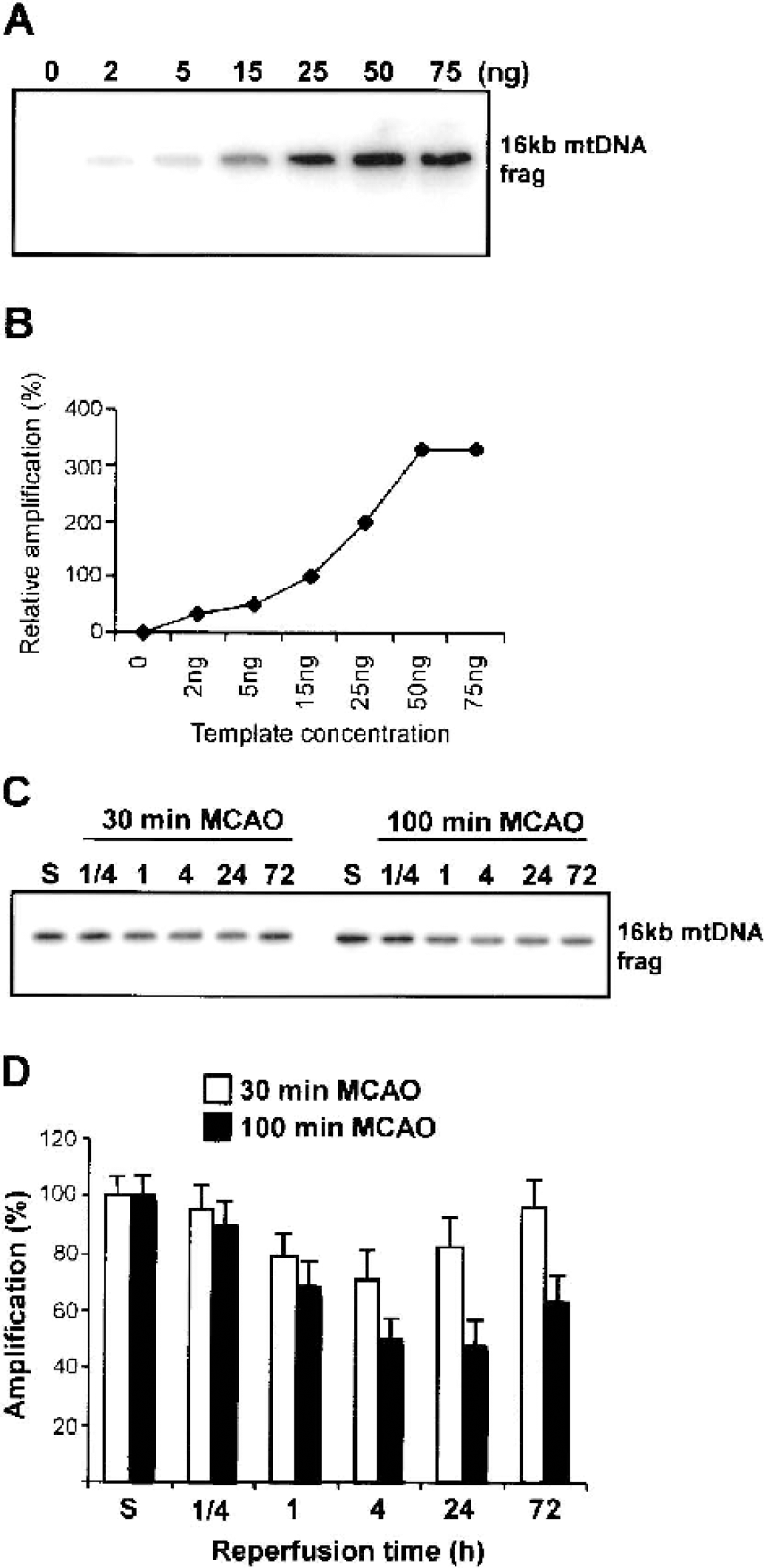
Detection of mitochondrial DNA (mtDNA) damage induced by cerebral ischemia assessed by quantitative polymerase chain reaction (QPCR) amplification.
Response of mitochondrial DNA base excision repair to focal cerebral ischemia
Several DNA lesions may result from oxidative DNA damage, including the deamination of cytosine to uracil and the oxidation of guanosine to 8-oxodG (Cadet et al., 2000; Croteau and Bohr, 1997), and these lesions are usually repaired by BER. 8-OxodG in mtDNA is a substrate for the mitochondrial isoform of oxoguanine DNA glycosylase 1 (mtOGG1 or mtODE). We therefore studied the ability of crude mitochondrial extracts from ischemic brain to incorporate [32P]-dGTP into oxidatively damaged pcDNA3.1 plasmids containing the oxidative adduct 8-oxodG. After 30-minute MCAO, BER of oxodG was upregulated at all time points examined (1/4 to 72 hours) (Figs. 3A and 3B). In contrast, 100-minute MCAO only briefly elevated BER activity from 15 minutes to 1 hour, with activity declining to below control levels thereafter (Figs. 3A and 3B). As a second measure of BER activity, we investigated the ability of mitochondrial extracts to repair a single U:G mismatch oligonucleotide (Fig. 4A). Increased mtDNA base excision repair was detected at all times after brief but not prolonged ischemia (Figs. 4B and 4C). Given the increase in BER activity, we decided to investigate the individual components of BER in more detail.
Mitochondrial uracil DNA glycosylase and apurinc/apyrimidinic endonuclease activity after ischemia
To assay UDG and APE activity in mitochondrial extracts, we designed a self-annealing oligonucleotide probe containing a single U:G mismatch at the 16th position, which is a substrate for UDG. To measure UDG and APE activity, the 32P-labeled probe was subject to BamHI restriction (Fig. 5A). UDG and APE activity was determined by the cleavage of the uracil containing oligonucleotide from 21 to 16 bases in length (Fig. 5B). Incision ability at the uracil site markedly increased after 30-minute MCAO at all time points examined. In contrast, UDG ability decreased after 100-minute MCAO from 1 to 72 hours after reperfusion (Fig. 5B).
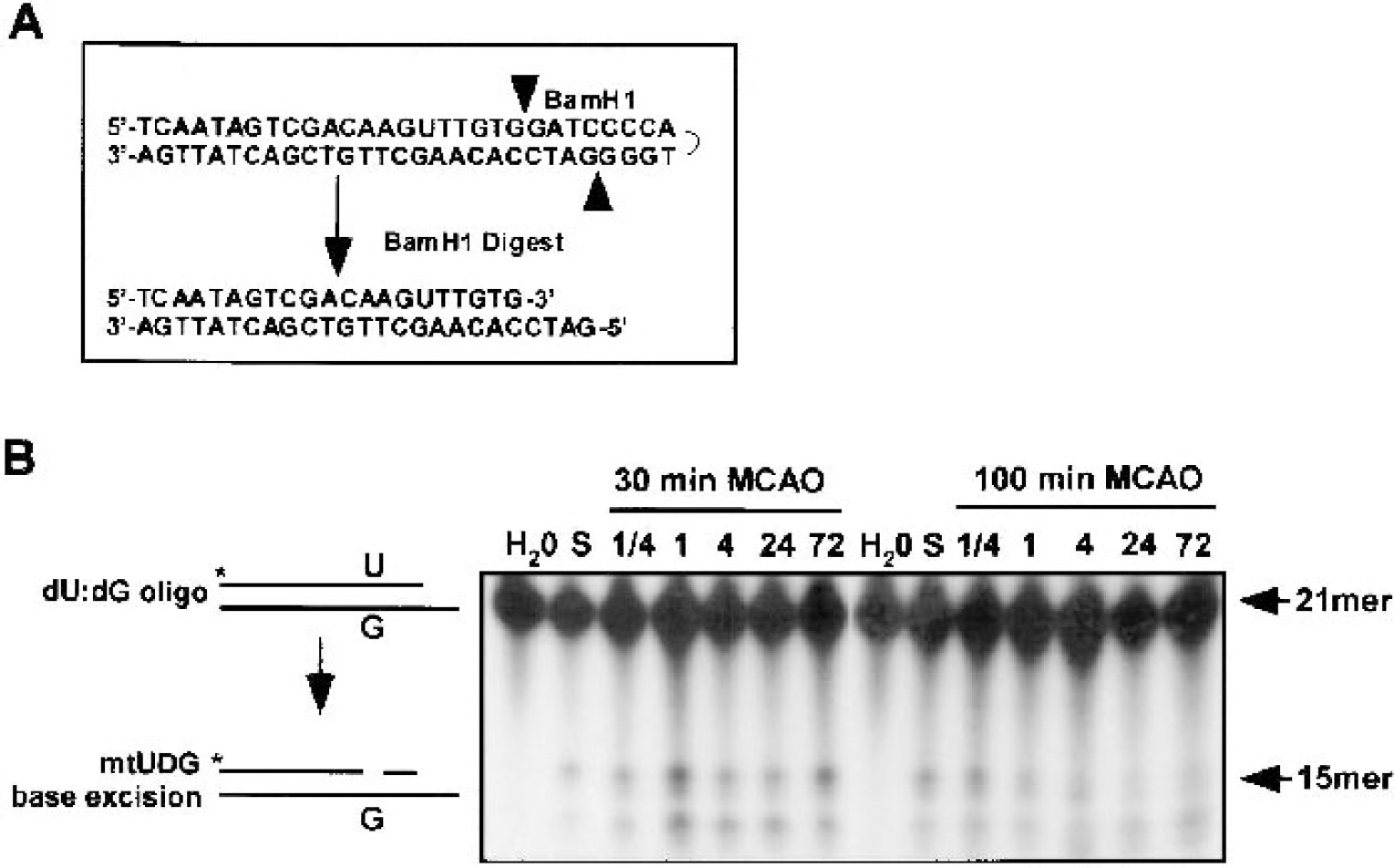
Detection of mitochondrial DNA UDG (MtUDG) and apurinic/apyrimidinic endonuclease activity.
To study the ability of mitochondrial uracil-DNA glycosylases (mtUDG) and mitochondrial AP endonuclease in crude mitochondrial extract, we used two different substrates: one has a single uracil lesion for which its incision requires both mtUDG and mitochondrial AP endonuclease (Fig. 5B), whereas the other substrate has one AP site for which incision only requires APE (Fig. 6B). Incision of these two oligonucleotide probes will release a 15-base fragment. The AP endonuclease assay produced comparable results to the UDG assay for 30-minute and 100-minute MCAO with elevated activity after brief (30-minute) MCAO and depressed activity in samples extracted from the 100-minute MCAO group (Fig. 6B).
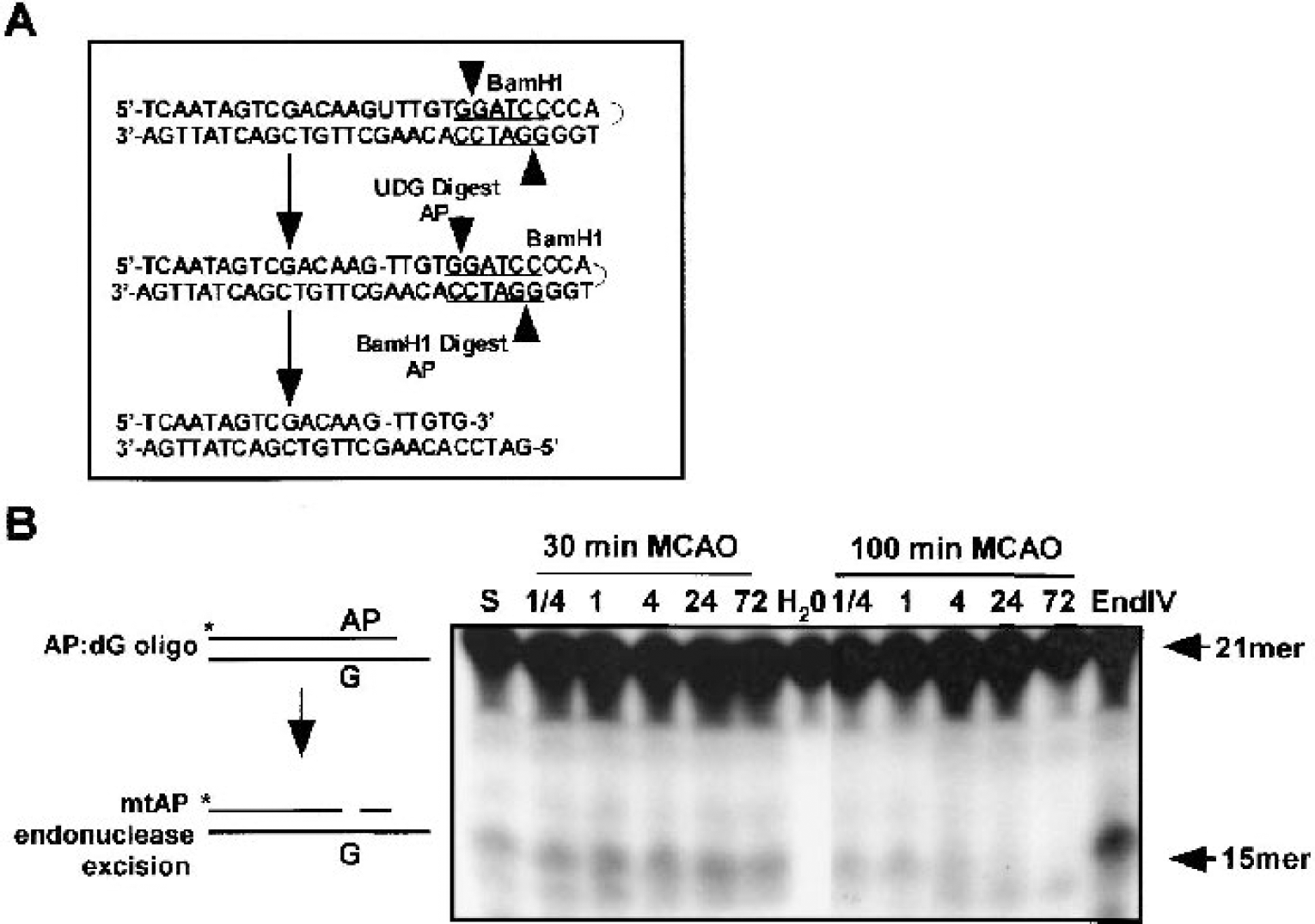
Detection of mitochondrial apurinic/apyrimidinic (mtAP) endonuclease activity.
Mitochondrial DNA polymerase activity after ischemia
DNA polymerization is the next step in the BER pathway for the repair of oxidative DNA lesions. The mitochondrial DNA polymerase-γ has been reported by several laboratories to be responsible for mtDNA replication and mtDNA repair. To determine the influence of brief or prolonged ischemia on mtDNA polymerase activity, we used a primer extension assay, whereby activation of DNA polymerase will result in a four-nucleotide extension (from 26mer to 30mer) as described previously (Chen et al., 2000) and as shown in Fig. 7A. After 30-minute MCAO, mtDNA polymerase activity significantly increased from 15 minutes to 72 hours (Fig. 7B) compared with levels in control brain. DNA polymerase I (large Klenow fragment) was included as a positive control (Fig 7B, lane 8). In contrast, mtDNA polymerase activity was only briefly elevated after 100-minute MCAO (at 15 minutes and 1 hour), declining to below control brain levels thereafter (Fig. 7B).
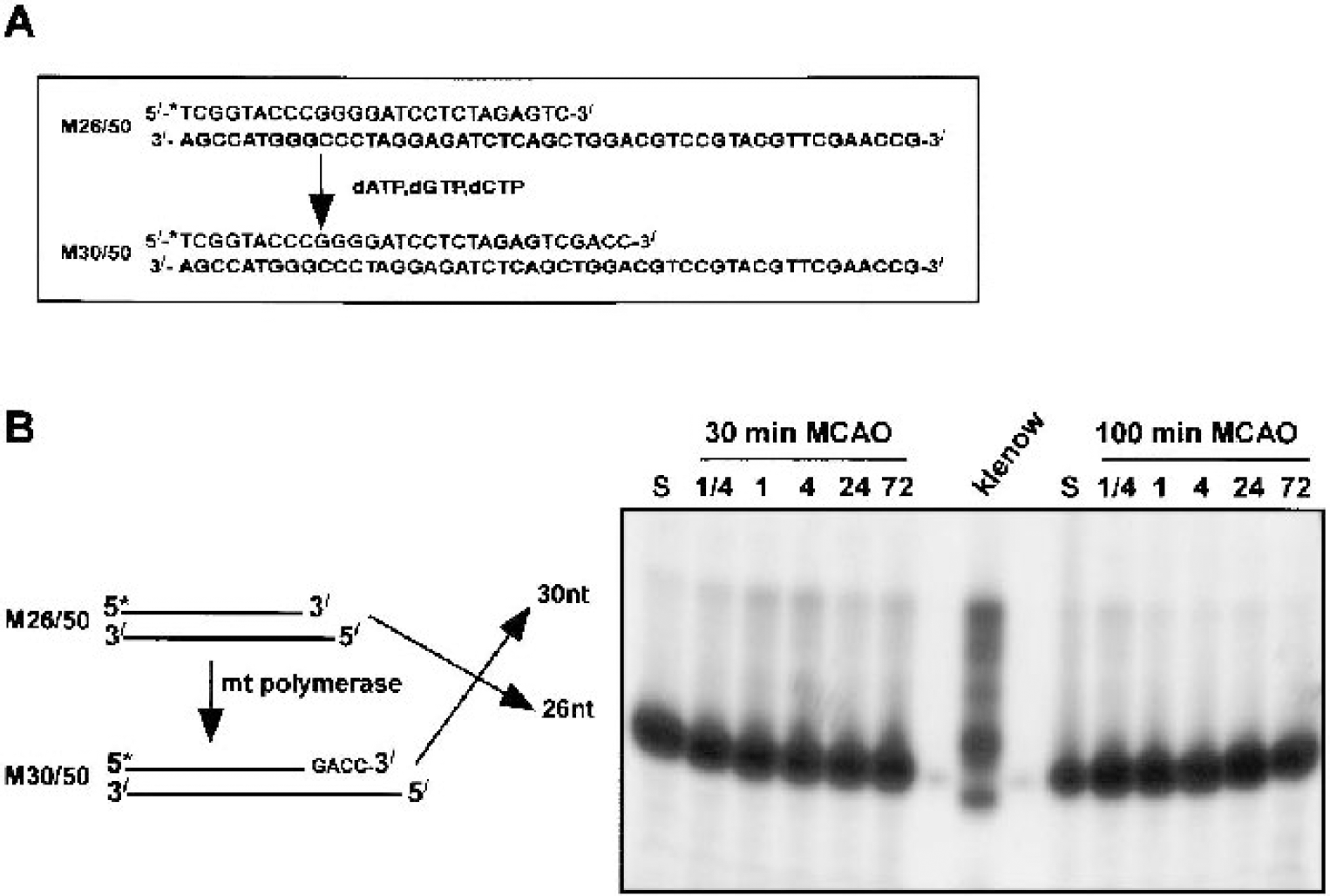
Detection of mitochondrial DNA polymerase activity.
Mitochondrial DNA ligase activity after focal cerebral ischemia
To finish the repair process, DNA ligase ligates the new DNA base to the adjacent base. Presently, we measured DNA ligase activity after 30-minute or 100-minute MCAO using an annealed dT18/polyA as a reaction substrate (Fig. 8). Briefly, increased ligase activity will ligate one or more labeled dT18 probes together, which can be resolved by electrophoresis as a 36-base product. We detected increased brain mtDNA ligase activity after 30-minute MCAO at all times examined (Fig. 8). After 100-minute MCAO, the mtDNA ligase ability was only induced from 15 minutes to 1 hour, decreasing to below control levels thereafter (Fig. 8).
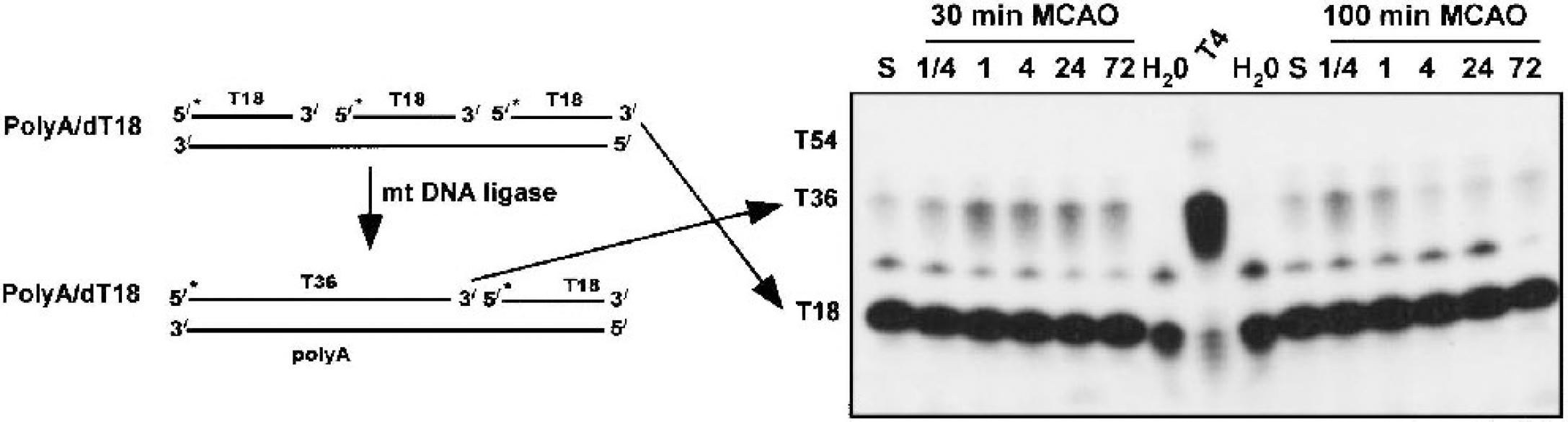
Detection of mitochondrial DNA (mtDNA) ligase assay.
DISCUSSION
In the present study, we examined the enzymes involved in the BER pathway of mitochondrial DNA (mtDNA) after mtDNA damage incurred by brief or prolonged focal cerebral ischemia. In these model systems, brief ischemia results in ischemic tolerance but prolonged ischemia evolves to infarction. Our data demonstrate that, after brief MCAO, mtDNA damage is mild and that there is early, prolonged, and enhanced activation of repair enzymes including 8-oxoguanine-DNA glycosylase (OGG), uracil DNA glycosylase, AP endonuclease, DNA polymerase, and DNA ligase. In contrast, after prolonged ischemia in which more severe mtDNA damage is incurred, the capacity of the BER pathway is only briefly elevated and remains ineffective or depressed thereafter. These data suggest that cellular recovery after ischemia may be dependent on the severity of the initial insult to mtDNA and the capacity of the mtDNA repair system to function. The mechanism of ischemic tolerance may extend to the ability of brain to repair mtDNA.
The mitochondrion is now well established as a critical instigator and reinforcement site in several cell death pathways, including those consequent on DNA damage (Green and Kroemer, 1998; Green, 2000; Green and Reed, 1998). Formation of oxidative DNA damage and inability to repair damage to mtDNA may underlie a number of neurodegenerative and aging-related diseases of the central nervous system (Green and Reed, 1998; Martin, 2001; Tritschler and Medori, 1993; Zhou and Elledge, 2000). DNA damage such as 8-oxodG, AP sites, and strand breaks may be caused by hydroxyl, peroxynitrite, and other damaging ROS that are generated during metabolic and pathologic processes (Cadet et al., 2000; Krokan et al., 2000). In the event of a reduction in effective mtDNA repair, accumulation of mtDNA damage and mutations in the mitochondrial genome lead to mitochondrial dysfunction and perhaps cell death (Green and Reed, 1998; Papa and Skulachev, 1997; Tritschler and Medori, 1993). Whereas antioxidants provide protection against potentially damaging oxidative stress, brain mitochondria have now been shown to possess the necessary enzyme machinery to repair damage to their DNA (Chen et al., 2002; Chen et al., 2000; Pinz and Bogenhagen, 1998), whereby oxidative DNA lesions such as 8-oxodG are replaced and ligated (Croteau et al., 1997; Croteau et al., 1999; de Souza-Pinto et al., 2001; Driggers et al., 1997; Pinz and Bogenhagen, 1998; Taffe et al., 1996).
This study was undertaken to examine the mtDNA BER pathway after focal cerebral ischemia and to test the hypothesis that an upgraded damage response mechanism in mtDNA could underlie, at least in part, a cell's ability to withstand future ischemic episodes (ischemic tolerance). Our preliminary study (Fig. 2) revealed that ischemia affected the integrity of the mitochondrial genome based on the inability of PCR to amplify the mitochondrial genome, supporting previous reports that ischemia rapidly induces DNA damage in brain (Cui et al., 1999, 2000; Liu et al., 1996; Tobita et al., 1995). We did not, however, attempt to identify the specific lesions in mtDNA that were induced by ischemia. Mitochondrial DNA damage incurred by brief focal ischemia quickly declined during the early reperfusion period, supporting previous studies on nuclear DNA that effective DNA repair mechanisms prevail when ischemic brain injury is mild (Cui et al., 2000; Liu et al., 1996; Nagayama et al., 2000b). Specifically, mitochondrial extracts from ischemic brain were capable of repairing an 8-oxodG lesion and a mismatched uracil, confirming previous studies that used control and aging brain mitochondria (Chen et al., 2000, 2002). Indeed, brief ischemia not only preserved mtDNA repair, but also in fact enhanced activity of these enzymatic pathways. In contrast, mtDNA damage and the activity of mtDNA repair enzymes could not be restored to nonischemic levels in the 24-hour and 72-hour reperfusion groups subject to prolonged (100-minute) focal cerebral ischemia (Figs. 2–7). These data confirm findings from nuclear DNA repair studies that a cell's capacity to repair mtDNA damage is dependent on the severity of the ischemic episode, with DNA left unrepaired when ischemia is dense or prolonged or both (Chen et al., 2001; Cui and Liu, 2001; Liu et al., 1996; Love, 1999; Martin, 2001; Moore et al., 2002; Sugawara et al., 2001).
Research on mtDNA repair mechanisms in ischemia remains limited, but evidence has accumulated for an effective mechanism for repair of oxidative lesions within mtDNA (Croteau et al., 1999). Our group and collaborators recently examined a number of key elements of the mtDNA BER pathway in brain (Chen et al., 2000, 2002), detecting the presence and function of BER enzymes capable of removing 8-oxodG, uracil, and AP sites as well as inducing DNA polymerase and ligase activities. Although we did not directly demonstrate the presence of the enzymes of the BER pathway in the mitochondrial fraction in our study, we can assume that our measured BER ability was of mitochondrial and not nuclear origin based on (1) fraction purity of our samples (i.e., the absence of any nuclear material [PARP] contamination in our tested fractions); and (2) our previous work supporting the presence of the BER in mitochondria (Chen et al., 2000, 2002).
Mechanisms for the targeting of each enzyme of the BER pathway to mitochondria have been examined. Previous reports identified nuclear and mitochondrial uracil-DNA glycosylases generated by alternative splicing and transcription from different positions in the UNG gene (Anderson and Friedberg, 1980; Nilsen et al., 1997). For example, it has been reported that there is an alternative last exon of the ogg1 gene that on differential splicing directs OGG1 to the nucleus (exon 7) or to the mitochondria (exon 8) (Croteau et al., 1997; Croteau and Bohr, 1997; de Souza-Pinto et al., 2001). The mtAP endonuclease is yet to be cloned. Extract characteristics and molecular weights, however, suggest it is a class II AP endonuclease (Pinz and Bogenhagen, 1998; Tomkinson et al., 1988). Although our experiments did not address which polymerase was present within mitochondrial fractions, previous reports would suggest that polymerase-γ is responsible for mtBER (Stierum et al., 1999). Previously, we demonstrated the presence of DNA ligase activity in mitochondrial extracts from rat brain (Chen et al., 2000). It has been reported that mtDNA ligase belongs to the DNA ligase III family, and mtDNA ligase has a mitochondrial targeting peptide at its N-terminal and its function is independent of PARP (Bogenhagen et al., 2001; Lakshmipathy and Campbell, 1999; Pinz and Bogenhagen, 1998; Tomkinson et al., 2001). Because we detected early and prolonged increases in DNA glycosylase, AP endonuclease, DNA polymerase, and DNA ligase activity in our mitochondrial fractions after brief ischemia, our data support the functional upregulation of the entire BER pathway within brain mitochondria after brief ischemia, with each enzyme activity being paralleled by each other.
The cellular phenotype underlying these changes is unclear, and we must consider that the changes in BER could be of nonneuronal origin, perhaps from glia. A glial proliferation-mediated origin, however, is unlikely because changes in mtBER enzyme activity were rapid and glial marker protein expression is in fact greater after 100-minute MCAO than after 30-minute MCAO (D. Henshall, personal communication, July 2002).
In contrast with brief focal cerebral ischemia, prolonged 100-minute MCAO that induced infarction within the ipsilateral cerebral cortex and striatum incurred more severe mtDNA damage. This is in keeping with findings on nuclear oxidative DNA damage after ischemia of varying severity (Nagayama et al., 2000b). Further, BER enzymes were only briefly (up to 1 hour after reperfusion) activated and declined thereafter, often to below control levels. Specifically, UDG, APE, DNA polymerase, and DNA ligase activity were all increased 1 hour after ischemia, but all were reduced to below control levels after this time point (see Results).
The ability of a cell to withstand ischemic injury likely depends in part on the induction of DNA repair mechanisms, as has been shown in the case of many key DNA repair enzymes such as PARP, which can lead to energy depletion and cell death or contribute to cell survival depending on the nature and severity of the ischemic challenge (Eliasson et al., 1997; Nagayama et al., 2000a). Although we did not address the underlying causes of the inability of mtDNA repair mechanisms to reactivate during reperfusion when ischemia was prolonged, this likely resides with a combination of prolonged energy failure, progressing mitochondrial breakdown, and the additional effects of reperfusion-induced free radical formation on restoration of blood flow. Further studies may better explain the nature of this failure to recover and the threshold at which injury repair capacity is breached.
In conclusion, brief focal cerebral ischemia induces a rapid, prolonged activation of the mtBER pathway in brain, which is enhanced over baseline. This prolonged activation profile is broadly in keeping with a possible underlying function in tolerance in which peak tolerance occurs 48 to 72 hours after preconditioning ischemia (Chen et al., 1996). Our data, however, do not entirely fulfill this criterion because the delay between initial and challenging (second) ischemic insult has previously been shown to require more than 24 hours to evolve. Our data suggest that the mtDNA BER pathway is often at peak activation before or during the 24-hour period after the ischemic challenge. Nevertheless, the BER pathway remained active 72 hours after ischemia, and further studies are necessary to more completely evaluate this mechanism. In contrast, a 100-minute ischemic episode induces marked oxidative damage to mitochondrial DNA and only a brief and likely ineffective activation of the BER pathway. These data confirm that a mitochondrial BER pathway is present and responsive to ischemic injury in brain, and suggest such DNA damage repair mechanisms may underlie the molecular switch between survival and death.